INTRODUCTION
Species of the red algal genus Pyropia (formerly Porphyra) are the most valuable seaweed crop worldwide. In 2017, global production of Pyropia spp. amounts to 2.5 Mt, for a value of USD 2.3 billion (http://www.fao.org/fishery/statistics/global-aquaculture-production).The culture of Pyropia is seriously affected by the oomycete Pythium porphyrae which causes the red rot disease (Gachon et al. 2010, Kim et al. 2014). The genus Pythium includes more than 250 species (Van der Plaats-Niterink 1981), most of which are soil-borne saprobes or facultative pathogens on diverse hosts, including plants, invertebrates and mammals (Van der Plaats-Niterink 1981, Lévesque and de Cock 2004). The first extensive molecular phylogeny defined 11 clades (A to K) encompassing all Pythium species (Lévesque and de Cock 2004). Clade K was later reassigned to the genus Phytopythium (de Cock et al. 2015), and there is now strong evidence supporting the lack of monophyly of Pythium sensu lato (Ascunce et al. 2017). Clade A includes P. porphyrae and the closely related Pythium chondricola which was initially isolated from decaying Chondrus. Both are most likely conspecific as P. chondricola has been mostly observed parasitizing Pyropia sp. (Lee et al. 2017, Qiu et al. 2019) and most classical molecular markers used to reconstruct oomycete phylogenies are strictly identical to P. porphyrae (Lee et al. 2015). Additional members of the same clade A include the soil-borne plant pathogen Pythium aphanidermatum (Lévesque and de Cock 2004) and pathogens occasionally reported on algal hosts (e.g., P. adhaerens) (Sparrow 1931). Genome sequences are available for Pythium species of agronomical interest (notably Pythium ultimum var. ultimum) (Lévesque et al. 2010, Ah-Fong et al. 2017), the emerging human pathogen Pythium insidiosum (Krajaejun et al. 2011, 2018), and the mycoparasite Pythium oligandrum because of its potential as a biocontrol agent (Horner et al. 2012, Berger et al. 2016, Kushwaha et al. 2017). Additional draft genomes have been generated for the plant-pathogenic Pythium ultimum var. sporangiferum, Pythium aphanidermatum, Pythium iwayamai, Pythium arrhenomanes, Pythium irregular, and Phytopythium vexans (Adhikari et al. 2013). Thus, the diversity of hosts and lifestyle amongst Pythium species make them good models for comparative genomics to study the evolution of pathogenesis amongst oomycetes. A decade ago, genome sequencing combined with high-throughput functional assays dawned an era of systematic characterization of pathogen virulence factors (generally termed effectors) secreted into the host apoplast or even addressed to the host cytoplasm to facilitate infection. The best studied oomycete effectors are small proteins sharing a conserved N-terminal RxLR signature (Morgan and Kamoun 2007). These so-called RxLR effectors are delivered into the host cytoplasm and the precise implication of the conserved RxLR signature in this translocation mechanism is highly debated (Ellis and Dodds 2011, Sun et al. 2013, Wawra et al. 2017). While RxLR effectors are expanded amongst Peronosporales, they are much less represented in other oomycete orders, notably absent from Pythium species (Adhikari et al. 2013), and the detection of RxLR-like proteins in other orders typically yields high numbers of false-positives (McGowan and Fitzpatrick 2017). Within the oomycete secretome, additional families of typical effectors include crinkling and necrosis factors (CRNs), necrosis inducing-proteins, elicitins, protease inhibitors and an array of peptidases and glycoside hydrolases (Jiang and Tyler 2012, McGowan and Fitzpatrick 2017).
As no genome or transcriptome data is yet available on P. porphyrae, we report here an expressed sequenced tag (EST) dataset generated using 454 sequencing. We provide a proteome and secretome prediction as well as a preliminary account of the genes potentially involved in P. porphyrae pathogenicity. We also performed gene clustering and de-novo approaches to assess if new pathogenicity-related proteins or host translocation mechanisms could be identified with this EST dataset. Finally, we also compared the P. porphyrae gene complement with that of known mammal and plant Pythium pathogens to get first molecular insights into the specialization towards a red algal host within the Pythium genus.
MATERIALS AND METHODS
Materials
A clonal strain of P. porphyrae was isolated from Porpyra sp. blades collected in Seochon (36°12′ N, 126°50′ E, Western Sea) and maintained at 6°C, under 15 μmol photons m−2 s−1 (10 h light, 14 h dark photoperiod) on corn meal agar plates. To generate free living hyphae of P. porphyrae, colonized agar plugs were transferred into liquid Arasaki B medium (Arasaki et al. 1968) and maintained for 7–10 days at 20°C in this suspension. Blades of Pyropia sp. host were grown in Modified Grund Medium (Klochkova et al. 2012) with constant aeration at 10°C under 30 μmol photons m−2 s−1 (16 h light, 8 h dark photoperiod). For the generation of EST libraries, P. porphyrae free living hyphae were inoculated into Pyropia sp. MGM cultures and harvested (three biological replicates) at 0, 4, and 30 h after inoculation. Mycelium was collected and thoroughly washed 4 times with sterile seawater to remove host debris and immediately flash frozen in liquid nitrogen.
RNA extraction and EST library assembly
Total RNA from P. porphyrae was isolated using TRI reagent (MRC, Los Angeles, CA, USA) according to the manufacturer’s protocol, treated enzymatically with DNase I (TaKaRa, Tokyo, Japan) to remove contaminant DNA, and purified with the Oligotex purification kit (Qiagen, Los Angeles, CA, USA). RNA quality was assessed both visually (1.2% formaldehyde gel electrophoresis) and using an Agilent 2100 Bioanalyzer (Agilent, Hambruk, Germany). 454 pyrosequencing library preparation and sequencing were carried out by GNCBio (Daejeon, Korea) using a GS Titanium Library Preparation kit (454 Life Sciences, Branford, CT, USA). cDNA libraries were validated using a High Sensitivity Chip on the Agilent 2100 Bioanalyzer, and emulsion PCR reactions were performed to enrich cDNA templates. Tagged libraries were combined in a picotiter plate and sequenced using a 454 Genome Sequencer FLX System (Roche, Los Angeles, CA, USA). The resulting Standard Flowgram Format file containing raw read data was used to filter low-quality reads (cut-off value of 40), primer and adapter sequences. Reads shorter than 50 bp were removed, and after pooling all conditions, the remaining dataset was de novo assembled using the GS De Novo Assembler software (Newbler v. 2.7; 454 Life Sciences). Datasets are available on the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) under the run accessions SRR11079882, SRR11079883, and SRR11079884.
Proteome prediction, clustering, and annotation of multigenic families
For the definition of open reading frames (ORFs), the EST dataset was locally blasted against the Pythium ultimum BR144 reference genome assembly (E-value cut-off, 1.0 E-5) and the resulting BlastX result file was fed into the OrfPredictor server (Min et al. 2005). The predicted proteome was further quality filtered with the CAST algorithm (Promponas et al. 2000), with a threshold set manually at 90, an all against all BlastP (E-value cut-off, 1.0 E-5) and a minimum similarity score of 20. The predicted ORF proteome was subjected to local all against all BlastP analysis (E-value threshold of 1.0 E-5) for the definition of gene clusters with TRIBE-MCL (Enright et al. 2002), using an inflation value of 2 (similar results were obtained with inflation values up to 7). A preliminary automated annotation of the proteome was performed using a combination of Blast2GO mapping (Conesa and Götz 2008), BlastP against the refseq_protein database (E-value cut-off, 1.0 E-6) and the PFAM batch server database (E-value cut-off, 1.0 E-5) (Punta et al. 2012). Homologues of genes potentially involved in pathogenicity were further manually curated, using a combination of reciprocal Blast analyses, database and literature searches.
De novo prediction of putative translocation motifs
Proteins targeted to the secretory pathway, i.e., either containing a signal peptide (SP) or a signal anchor (SA) were identified with HECTAR (Gschloessl et al. 2008). The SP cleavage sites were predicted with PredSL (Petsalaki et al. 2006). Gene clusters containing secreted (SP) or signal anchor-containing (SA) proteins were defined based on the TRIBE-MCL clustering described above. Similar to the strategy of Saunders et al. (2012), a de novo motif search was performed with MEME (Bailey et al. 2009) to identify ab initio potential RxLR-type host translocation motifs. Since our EST sequences might be truncated at the 5′-terminus, we chose not to impose any a priori positional constraint. MEME was run on the 194 SP proteins contained in the 43 clusters encompassing more than 4 records. The program was set to report the 25 most robust motifs of 4 to 40 amino acids, occurring zero or once per sequence. The resulting MEME motifs were visually scored according to the following criteria: (1) how many times the motifs are present; (2) their dispersion of their position along the protein sequence sites; and (3) whether they were found across different tribes (as would be expected for a conserved translocation motif). Because subcellular targeting algorithms sometimes struggle to discriminate SP from SA, we also ran as a control the same analysis on all SA-containing clusters
Sequence similarity networks for oomycete elicitin and elicitin-like genes
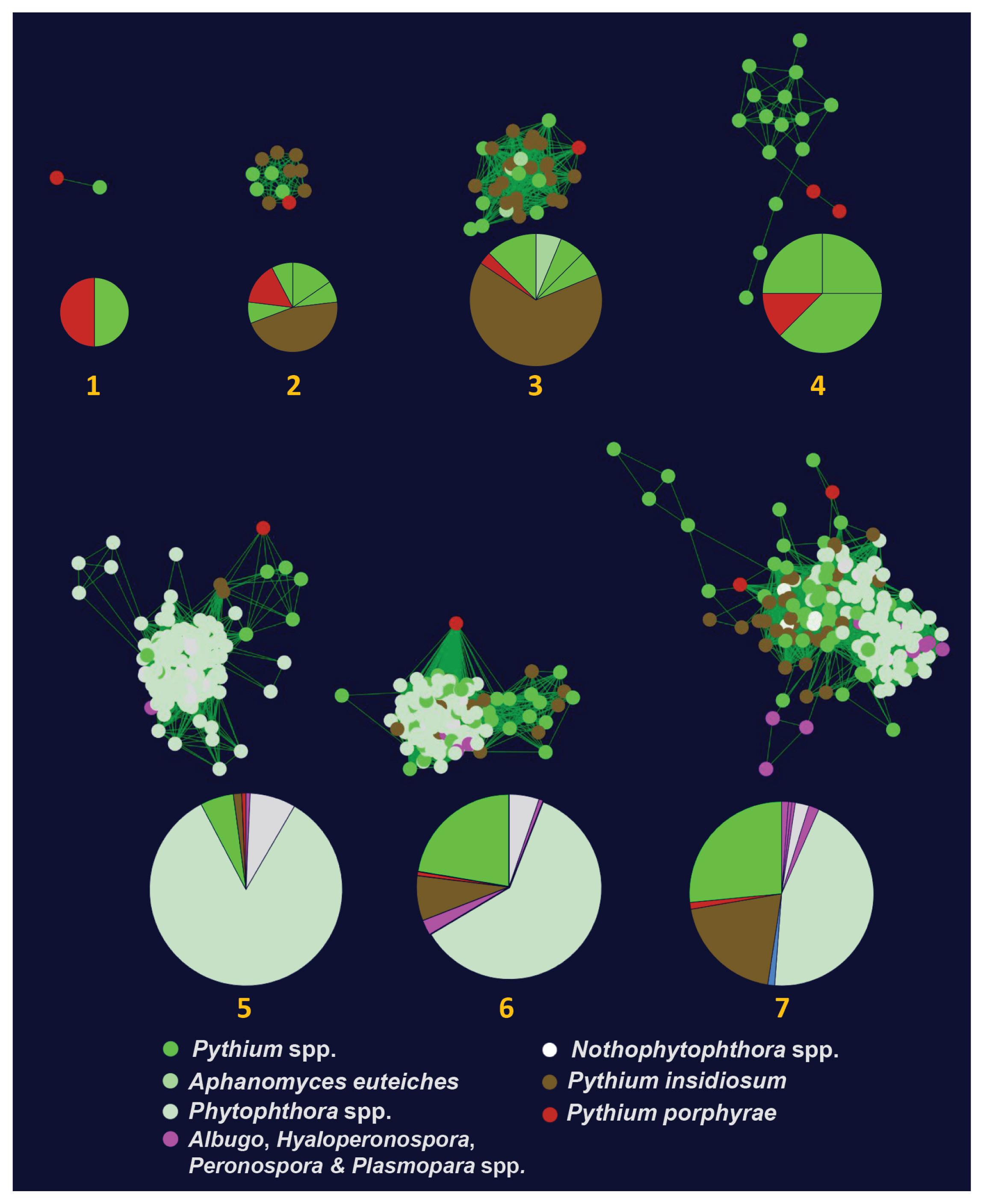
P. porphyrae elicitins and elicitin-like genes were identified using similarity and PFAM searches, aligned in CLC Genomics Workbench and the alignment was visually inspected and corrected for misalignments. The resulting alignment was trimmed, exported in Clustal (.aln) format and used to generate an hmm model with HMMER3 (Eddy 2011). The resulting hmm model was used to screen all Pythium datasets mentioned in the section below (E-value cut-off, 1.0 E-5), yielding 321 Pythium sp. proteins. Additionally, 1,931 oomycete proteins containing elicitin or elicitin-like domains were obtained from the NCBI by keyword search, as well as 31 proteins annotated as elicitins or elicitin-like on the AphanoDB repository (https://www.polebio.lrsv.ups-tlse.fr/aphanoDB/). All 2,219 proteins were loaded on the EFI-EST (Enzyme Function Initiative-Enzyme Similarity Tool) online server (Gerlt et al. 2015) and used for the generation of sequence similarity networks (SSNs) using default parameters for edge calculation. To account for diversity of domains and protein length present in the dataset, an edge score threshold of 7 (equivalent to an E-value cut-off, 1.0 E-7) was selected for the definition of similarity clusters. Resulting SSNs were visualized and analyzed in Cytoscape (Demchak et al. 2014).
Clustering with additional known Pythium genomes
To define P. porphyrae specific proteome, the genomes of P. ultimum var. ultimum, P. ultimum var. sporangiferum, P. iwayamai, P. aphanidermatum, P. irregulare, P. arrhenomanes, Phytopythium vexans (formerly Pythium vexans) and the genome of the animal pathogen P. insidiosum were obtained from the Pythium Genome Database (http://pythium.plantbiology.msu.edu/) and the NCBI (www.ncbi.nlm.nih.gov/genome/34156), respectively. Gene clustering was performed through the get-homologue software (Contreras-Moreira and Vinuesa 2013) using the implemented OrthoMCL (option -M) algorithm to report all possible gene clusters, including species-specific clusters (parameter -t = 0). A Pangenome Matrix was computed (compare_clusters.pl script available through get_homologues) and used to define the P. porphyrae specific proteome.
RESULTS
Assembly and initial annotation of the Pythium porphyrae gene repertoire
A clonal strain of P. porphyrae was isolated in 2012 from infected Pyropia sp. blades growing in Seochon (36°12′ N, 126°50′ E). Clear symptoms of red rot disease could be observed, starting from small red patches on the blades where fungal spores germinated. Areas of dead host tissue spread and ultimately lead to the disintegration of the entire blades (Fig. 1A). Classical optical microscopy confirmed the typical intracellular growth of P. porphyrae through host algal cells (Fig. 1B). Three EST libraries were generated from P. porphyrae after 0, 4, and 30 h of exposure to Pyropia blades, and corresponded to 630,388, 667,490, and 575,530 reads, respectively. The EST corresponding to the 18S ribosomal RNA (EPT001TT0381C007649) was 100% identical to known P. porphyrae sequences (AY598673 and JQ898472), thus validating the taxonomic assignment of this isolate as P. porphyrae (Fig. 1C). An overview of our EST analysis strategy is illustrated in Fig. 2. The reference transcriptome pooling all ESTs libraries contained 10,819 unigenes, representing 11.1 Mb of non-redundant sequence data. The corresponding partial proteome extracted by OrfPredictor consisted of 10,807 predicted ORFs. Further filtering (i.e., excluding sequences under 41 amino acid, and CAST quality filtering) led to a final ORF dataset of 10,554 sequences (hereafter referred to as the “P. porphyrae proteome”), of which 77% (8,150 proteins) could be assigned to PFAM hits (Supplementary Table S1). Assessment of completeness using the BUSCO software (Simão et al. 2015) reported 89.9% of 429 core eukaryotic genes (Supplementary Table S2). As can be expected for an EST dataset, a significant proportion of the detected genes (14%) were fragmented, and the overall completeness was less than for most fully sequenced Pythium. For the definition of multigenic gene families, TRIBE-MCL clustering retrieved 1,216 gene clusters of two or more ORFs (Fig. 3, Supplementary Table S1).
Annotation of putative pathogenicity-related genes
Over the last decades, several protein families have been implicated in oomycete pathogenicity. The most studied families are listed in Table 1 together with an account of their size in P. ultimum and Phytophthora infestans. Among these, we manually curated the potential P. porphyrae orthologues with a focus on RxLR effectors, crinklers (CRN), elicitin / elicitin-like proteins, protease inhibitors, cellulose-binding elicitor lectins (CBELs), and other proteins that could relate to cell-wall binding or hydrolysis. A full list of the genes manually inspected in this study is provided in Supplementary Table S3.
RxLR effectors and crinklers
Despite extensive searches with BLAST or regular expressions, no expanded RxLR family in P. porphyrae could be identified. Only two proteins, EPT001TT0381C005379 and EPT001TT0381C004327, displayed significant homology to putative RxLR-like proteins conserved across Peronosporales and Pythiales when blasted against the nr database, with the highest similarity to P. insidiosum (57.7% to GAY02831.1 and 60.4% to GAX97019.1, respectively). A SP could be detected for both proteins but only EPT001TT0381C004327 carried an RxLR motif. When blasted against the broad spectrum of RxLR-like proteins defined by McGowan & Fitzpatrick (McGowan and Fitzpatrick 2017), EPT001TT0381C005379 was most similar to a Phytophthora cryptogea (PHCR|05525, 58.9% identity) protein that did not cluster with any other omycete species. On the contrary, EPT001TT0381C004327 was most similar to a P. arrhenomanes protein (PPYAR|006221, 60.9% identity) belonging to a conserved multispecies cluster of putative RxLR-like proteins (https://oomycetes.github.io/rxlrs.html). We also identified 9 sequences homologous to the P. ultimum YxSL[RK] proteins, which was hypothesized as a new family of RxLR-like candidate effectors (Lévesque et al. 2010). As in P. ultimum, the gene cluster detected for P. porphyrae family was especially rich in secreted proteins (eight out of nine members). However, the YxSL[RK] motif was only poorly conserved, replaced by Y[KS][TS]L[RK] in P. porphyrae, and in one instance interrupted by an insertion (Supplementary Fig. S1).
In total, 13 CRNs were identified by similarity search with other known oomycete CRNs (Supplementary Table S3). All contigs that spanned the protein N-terminus had an N-terminal LxLYLAR consensus (Supplementary Fig. S2, red triangles) clearly related to the P. ultimum LxLYLAR/K motif (Lévesque et al. 2010). The LxLYLAR motif was followed by two conserved WL residues (Supplementary Fig. S2, yellow triangles) and a stretch of variable amino acids. As in P. infestans, a highly conserved HVLVxxP motif (Supplementary Fig. S2, orange triangles) linked the conserved N-terminus to the variable C-terminus. Eleven of the 13 identified P. porphyrae CRNs contain a domain already reported in a P. infestans CRN, including the DC domain, which was shown to stimulate cell death when expressed in planta (Haas et al. 2009).
Cellulose-binding proteins and cell wall hydrolases
While looking for determinants of pathogenicity on red algae, we searched for enzymes that might be specifically involved in the degradation of red algal carbohydrates, especially sulphated polysaccharides. We found about twenty sulphatases and sulphate permeases that were well conserved with Phytophthora, and therefore likely irrelevant in the context of red algal cell wall degradation (data not shown). Extensive searches for enzymes bearing homology to bacterial agarases, carrageenases, and porphyranases (families GH16, 50, 86, 96, and 118) also remained unsuccessful. On the other hand, we identified three cellulases (EPT001TT0381C001561, EPT001TT0381C001658, EPT001TT0381C001964), as well as a putative cutinase (EPT001TT0381C004750) most closely related to Phytophthora cutinases. Finally, we found seven CBEL-like (cellulose-binding elicitor lectins) proteins carrying both carbohydrate binding modules (CBMs) and PAN/APPLE domains (Supplementary Table S3). The gene cluster containing these CBEL-like proteins contained 11 additional proteins with PAN/APPLE domains alone or in combination with other domains (Supplementary Fig. S3).
Elicitins
We identified 26 elicitin-like proteins in P. porphyrae (Supplementary Table S3). Eighteen proteins had a predicted SP, whereas the lack thereof in the others was likely due to incomplete coverage of the N-termini. We aligned and visually inspected elicitin-like domain structure of P. porphyrae, validating the conservation and spacing of the 6 cysteine residues for 22 proteins (Supplementary Table S3). No known C-terminal domains could be detected on any of the 26 proteins. An hmm model derived from P. porphyrae elicitins was used to screen all putative elicitin-like proteins from the genomes of 8 additional Pythium species and coupled to all available elicitin-like proteins from public databases to generate a SSN (see methods). Only 9 P. porphyrae elicitins were assigned to 7 oomycete elicitin clusters (Fig. 4, Supplementary Fig. S3) using an EFI-EST edge score threshold of 7. Note that increasing this threshold to 10 and above abolished most clustering of the P. porphyrae proteins, thus reflecting the overall divergence of this elicitin-like repertoire. Three clusters were specific to the Pythiales (Clusters 1, 2, 4 on Fig. 4) while the others overlapped the Peronosporales and / or Albuginales. The 17 remaining P. porphyrae elicitins could not relate to any known oomycete elicitins (Supplementary Fig. S3).
Ab initio prediction of secreted candidate pathogenicity effectors
To detect putative novel pathogenicity effectors, we screened the P. porphyrae secretome for candidate translocation motifs. Supplementary Table S4 contains the results of the subcellular targeting software HECTAR. 918 ORFs (8.7% of the proteome) were predicted to contain a SP and 333 to contain a SA (3.2% of the proteome). This result is in the range of other known oomycetes (e.g., 7.7 and 7.5% of secreted proteins in Phytophthora sojae and P. ramorum) (Tyler et al. 2006), but higher than predicted in P. ultimum (4.9%) (Lévesque et al. 2010). The PFAM analysis retrieved 1,476 and 558 hits, mapped against 724 SP and 284 SA proteins (78.9 and 85.3% of all SP and SA ORFs, respectively) (Supplementary Table S5). We defined clusters of secreted proteins following the original TRIBE-MCL clustering (Fig. 3) and ran MEME on these subsets. Generally, amino-acid motifs found in the secretome were highly tribe-dependent, and essential characteristic of specific protein families. Despite our extensive unbiased searches, we have been unable to identify any known or novel candidate translocation motif in P. porphyrae.
Overlap of Pythium porphyrae gene repertoire with other Pythium species
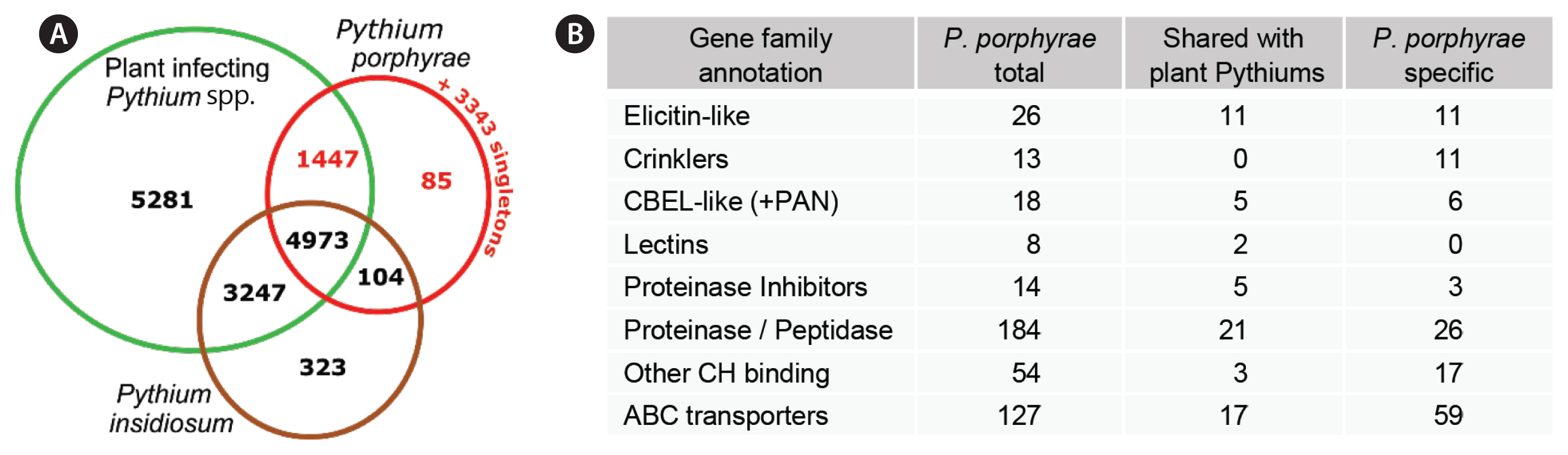
To identify candidate genes that may account for the algal host specificity, we used a standard OrthoMCL clustering to compare our gene repertoire to the proteomes of 7 plant-associated Pythium species (analyzed by Lévesque et al. 2010) and to the proteome of the animal pathogen P. insidiosum (see methods). A core proteome of 4,973 clusters (5,299 genes) was shared between all included Pythium species (Fig. 5A). Of the 6,609 P. porphyrae clusters, 104 were shared only between P. porphyrae and P. insidiosum, while a total of 1,447 clusters (representing 1,507 P. porphyrae proteins) were shared with at least one plant-pathogenic Pythium species and not detected in P. insidiosum. Altogether, 34% of the proteome was specific to P. porphyrae, with 200 proteins in clusters and 3,443 singletons. Interestingly, numerous putative pathogenesis-related candidates could be found in the specific P. porphyrae proteome defined by OrthoMCL (Fig. 5B), notably 11 elicitin-like and the majority of CRNs. For CBEL-like proteins, the association of CBM and PAN/APPLE was not specific to phytopathogenic Pythium species as homologs were also detected in P. insidiosum (Supplementary Fig. S3).
DISCUSSION
Here we present a first account of the gene repertoire of Pythium porphyrae the genome of which has not been sequenced to date. Thus, our analysis of this EST dataset provides an interesting insight into the molecular determinants of pathogenicity of the most destructive disease-causing organism in Pyropia farms.
The Pythium porphyrae gene repertoire conforms to expectations
In terms of gene content, P. porphyrae mostly conforms to the trends described in other Pythium species and across oomycetes (Table 1). We detected all the classic families of proteins reported in other Pythium species as candidate pathogenicity proteins, including CRNs, Elicitings, CBELs, protease inhibitors. Despite extensive searches, and consistent with other Pythium species, we failed to evidence any expanded RxLR effector family or new candidate translocation motifs with similar structural properties (Lévesque et al. 2010, Horner et al. 2012, McGowan and Fitzpatrick 2017, Krajaejun et al. 2018). Finally, only one of the 14 CRNs identified in P. porphyrae is predicted as secreted. Despite the caveat that some of our sequences may not be full length, this resonates with the earlier finding that only two among the 26 P. ultimum CRN proteins contained a SP (Lévesque et al. 2010). Our observations suggest that the primary function of Pythium CRNs may not be to act as virulence effectors translocated into the cytoplasm of their host. Altogether, and similar to other described Pythium species, P. porphyrae pathogenicity is unlikely to reside in fast-evolving, expanded families of host-translocated pathogenicity effectors similar to those described in Phytophthora and other (hemi)biotroph oomycete pathogens.
The enzymatic arsenal of a reformed plant pathogen?
In our attempts to identify molecular determinants of host specificity, we were unable to identify any degradative enzyme that would specifically target red algal polysaccharides. It is worth noting that while such enzymes have been identified in prokaryotes (Fu and Kim 2010, Schultz-Johansen et al. 2018), eukaryotic counterparts have yet to be characterized. Thus, we cannot exclude the possibility that some P. porphyrae specific hydrolases reported here may act as red algal degradation enzymes.
Although P. porphyrae has not been found on any other organism than Pyropia, a recent report demonstrated that several plant species inoculated with P. porphyrae develop symptoms including root rot and, occasionally, elicit a hypersensitive response (Klochkova et al. 2016). Here, we evidenced a cutinase, an enzyme specialized in the degradation of plant cuticles. Several cellulases are also present, although cellulose is notably absent from the gametophytic (i.e., blade) stage of Pyropia infected by P. porphyrae (Mukai et al. 1981). These elements suggest that P. porphyrae has evolved from and retained the metabolic capacity of a saprophyte or pathogen exploiting terrestrial plants. We hypothesize that the plant defence response or symptoms triggered by P. porphyrae lab-controlled inoculation results from the detection by the hosts of conserved microbial signatures such as the elicitin-like and CBEL-like proteins reported in this study.
In conclusion, we only identified gene families shared with known oomycete parasites. Although our comparative approaches suggest some degree of specificity for several classic pathogenicity proteins of P. porphyrae (Fig. 5B), the molecular determinants of adaptation to red algal hosts remain elusive. Savory et al. (2018) speculated that the broad host range of P. aphanidermatum, the closest relative of P. porphyrae within clade A, could be linked with a striking expansion of substrate range for a transporter acquired through horizontal gene transfer. Using the gene of P. aphanidermatum (pag1_scaffold_175-fgenesh-gene-0.5) as bait, we could not to retrieve a credible ortholog for P. porphyrae.
Keeping in mind the inherent limitations of an EST dataset (completeness, truncated transcripts) for gene clustering, we hope that accessing the full genomic context and detailed gene expression patterns in a time course of infection will improve our understanding the diversity and functions of P. porphyrae pathogenicity determinants.