INTRODUCTION
The green microalga Chlamydomonas reinhardtii is a widely used unicellular model organism for the study of flagellar structure and function, genetics of basal bodies, chloroplast biogenesis, photosynthesis, light perception, cell-cell recognition, and cell cycle control (Harris 2001). Analysis of the genome of C. reinhardtii reveals the evolution of key animal and plant functions (Merchant et al. 2007). It is also used as a model for the study of biofuel production (Wang et al. 2009, Moellering and Benning 2010, Work et al. 2010).
Insertional mutagenesis is a popular methodology for screen and isolation of mutant strains with specific phenotypes in C. reinhardtii. This is because the known insert-DNA sequences facilitate the identification of the insertion site in isolated mutant strains. A number of methods such as plasmid rescue (Tam and Lefebvre 1993), inverse polymerase chain reaction (inverse-PCR) (Yoshioka et al. 2004), and thermal asymmetric interlaced polymerase chain reaction (Pollock et al. 2003) are developed for determination of genomic sequences flanking to known insert-DNA fragments. Inserts that contain antibiotic resistant marker genes such as AphVIII (Sizova et al. 2001) and Ble (Lumbreras et al. 1998) against paramomycin and bleomycin (respectively) are frequently adopted for mutagenesis in wild type cells. To ensure antibiotic resistance, marker genes are often driven by robust promoters such as RBCS promoter or PSAD promoter (Lumbreras et al. 1998, Sizova et al. 2001). In most cases, insertional mutagenesis creates loss-of-function mutations or null mutations.
Sub-saturation screening using insertional mutations for genes involved in photosynthesis, nitrate assimilation, and acclimation to sulfur deprivation have provided tremendous insight into mechanisms regulating these processes and made resources available to the study of respective processes in C. reinhardtii (Dent et al. 2005, González-Ballester et al. 2005, Pollock et al. 2005). The amenability of the C. reinhardtii genome makes it suitable as green cell-factories to produce value-added metabolites and heterologous proteins (León-Bañares et al. 2004). However, the biomass yield of C. reinhardtii is relatively low when compared to some industrial green microalgal strains. To improve the biomass productivity, insertional mutant strains with reduced pigment or truncated antenna are found to show increased biomass yield under high but not low illumination conditions (Polle et al. 2003). This is because truncated antenna cells on surface of photobioreactors shield less light so that cells under the surface receive more light than wild type cells under high illumination conditions (Polle et al. 2003). On the other hand, biomass yield of truncated antenna cells is similar to that of wild type because no significant amount of lights is shielded by cells on surface (Polle et al. 2003).
To explore the possibility for having mutants exhibiting high biomass yield under low-illumination conditions in C. reinhardtii, we performed forward genetic screening for insertional mutant strains exhibiting high cell-density (HCD) phenotypes from a pool of >1,000 individual insertional mutant strains as a proof-of-principal study. Through 30 cycles of continuous subcultures, we have isolated 3 non-allelic strains exhibiting HCD phenotypes under low-illumination growth conditions. HCD strains show no apparent decrease of chlorophyll content. Inverse-PCR analysis identified the insertion site in 2 out of 3 HCD mutants, namely the HCD1 and HCD3. In both cases, genes associated with mitochondrial function at the insertion sites are found to be transcriptionally induced. Together, our results show that HCD strains can be enriched through continuous subcultures under low-illuminations. During the preparation of this manuscript, Cheng et al. (2017) have published a large-scale screening of 150 thousand insertional mutants. This provides the great resources for near-saturation screening of HCD mutant cells using the continuous subculture methodology in the future. We propose that near-saturation screening against the 150 thousand insertional mutants using this continuous subculture methodology will allow gaining insight into molecular mechanisms for regulation of cell-culture density in C. reinhardtii and possibly other green algae.
MATERIALS AND METHODS
Strains, plasmids, and culture growth conditions
The C. reinhardtii strain CC503 and plasmid pSP124S are acquired from the Chlamy center (http://www.chlamy.org). CC503 is cultivated in high salt (HS) (Sueoka 1960) and TAP media (Harris 1989) under autotrophic and heterotrophic growth conditions (respectively) of illumination at 50 μmol photon m−2 s−1, shaking at 70–90 rpm, temperature at 25°C, and bubbling with 2–3% CO2 at ~100 mL min−1 as indicated when appropriate. Cell density is determined by colorimetric method (optical density [OD] at 750 nm wavelength) or gravimetric method (cell dry weight). Continuous subcultures of stationary-phase cells were carried out by taking culture (3 days, ~1.4 OD) as inoculum to seed the fresh medium (0 day, ~0.15 OD) and continue cycles of subcultures.
Analysis of chlorophyll contents
To determine chlorophyll content, cells from 2 mL culture were harvested by centrifugation. The resulting cells were resuspended in 96% ethanol and broken in the glass-bead beater FastPrep homogenizer (MP Biomedicals, Solon, OH, USA). After incubation with ethanol for 2 h at 4°C, cell debris was cleared by centrifugation. The resulting supernatant was subjected to OD measurement at the wavelength of 645 and 663 nm, with the 96% ethanol as blank. Total chlorophyll content (i.e., chl a and b) was estimated using a formula C (mg L−1) = 20.2OD645 + 8.05OD663 (Sartory and Grobbelaar 1984) and was converted to percent of cell dry weight.
Analysis of cell diameters and numbers
Cell diameters and numbers were determined by using a particle analyzer (Multisizer 3; Beckman Coulter Life Sciences, Indianapolis, IN, USA) following the manufacturer’s instruction. In brief, cells were collected from fresh culture by centrifugation at 2,500 rpm, washed with phosphate buffered saline (PBS) and resuspended with PBS to the initial volume. PBS washed cells were mixed with electrolyte (Beckman Coulter Life Sciences) with a ratio of 1 : 19 v/v. Cells in electrolyte were subjected to diameter and number analysis using the Multisizer 3. Each analysis in triplicate was performed based on 100 μL of cells in electrolyte.
Analysis of mitochondrial contents
To investigate mitochondrial content in HCD1 and HCD3, a fluorescence microscopic analysis was performed using Mitotracker Green FM (Molecular Probes Inc., Eugene, OR, USA). Cells were stained with Mitotracker Green FM at a final concentration of 0.1 g mL−1 for 30 min at 25°C. The stained cells were observed under a fluorescence microscope (BX53; Olympus Co., Tokyo, Japan) and images were taken using a CCD camera (DP73; Olympus Co.). Fluorescence intensity of cells was determined using ImageJ (http://imagej.nih.gov).
Insertional mutant library construction
To screen for insertional mutant strains with HCD phenotype, insertional mutants were constructed. Briefly, CC503 cultured in TAP medium were harvested and transformed with a 1.17 kb Ble gene fragment which was PCR-amplified from the plasmid pSP124S (Lumbreras et al. 1998) using the primers: Ble_1F (5′-TGGAAGCT TAAATGCCAGAAGGAGCGCAGCC-3′) and Ble_1174R (5′-CCGAGCTCAGCTTCAAATACGCCCAGCCCG-3′) (for nucleotide sequence of PCR fragment of the Ble expression cassette) (see Supplementary Fig. S1). Transformation was performed via electroporation as previously described (Shimogawara et al. 1998). The transformants were selected on TAP agar plates supplemented with 2.5 μg mL−1 of zeocin (Invitrogen, Thermo Fisher Scientific Co., Waltham, MA, USA).
Genetic screening for HCD insertional strains through continuous subcultures
Over 1,000 transformed colonies were picked and pooled into a flask containing HS medium (Sueoka 1960) supplemented with 2.5 μg mL−1 zeocin and grown photoautotrophically to OD750 > 1.0 under continuous illumination of 50 μmol photons m−2 s−1 and 2% CO2 bubbling. The pool of mutants were subsequently used to seed three flasks to OD750 = ~0.15. Continuous subcultures of stationary-phase cells were carried out for 30 cycles. Cell samples of the initial pool of mutant strains (0 subculture), the 10th, 20th, 30th cycles of subcultures were subjected to restriction fragment length polymorphism (RFLP) analysis.
RFLP analysis
To investigate growth of various insertional mutant strains during continuous subcultures, RFLP analysis of Ble sequences was performed. Briefly, cells were lysed using CTAB buffer (2% w/v cetyltrimethyl ammonium bromide, 100 mM Tris-HCl pH 6, 50 mM ethylenediaminetetraacetic acid pH 7, 1.5 M NaCl, 50 mM dithiothreitol) and genomic DNA molecules were subsequently extracted with phenol : chloroform. Probe for detection of Ble sequences was PCR amplified using primers Ble_E2_F (5′-TGGCCAAGCTGACCAGCGCCGTTCC-3′) and Ble_E3_R (5′-CCTCCGACCACTCGGCGTACAGC-3′) on plasmid pSP124S (Lumbreras et al. 1998) as template (DIG Probe Synthesis Kit; Roche, Basel, Switzerland). Southern blotting analyses were performed according to manufacturer’s instruction with buffers and solutions provided. The α-DIG-AP was diluted 1 : 15,000 and washing buffer composition was modified to 0.1 M maleic acid, 3 M NaCl, 0.3% Tween 20, pH 8.0 to optimise the DIG detection step (Engler-Blum et al. 1993).
Inverse-PCR analysis for identification of genomic sequences flanking to insertion locus
To identify the sequences flanking to the Ble insert, inverse-PCR methodology (Ochman et al. 1988) was utilized. Briefly, mutant genomic DNA was subjected to a single restriction enzyme digestion using one of restriction enzymes including but not limited to NheI, NdeI, and PstI. T4 DNA ligase (Life Technologies, Thermo Fisher Scientific Co.) was used to circularize the digested DNA. The resulting circularized DNA was used as template for PCR amplification of genomic DNA flanking to Ble insert using primers Ble_E2_R (5′-CCGGAACG GCGCTGGTCAGCTTG-3′) and Ble_E3_F (5′-CTGCGT GCACTTCGTGGCCGAGG-3′). Flanking genomic DNA sequences derived from the inversely amplified DNA fragment were subjected to BLAST analysis against the genomic sequences of C. reinhardtii using the Phytozome tools (https://phytozome.jgi.doe.gov). Insertion locus in HCD1 and HCD3 was further validated using sequence-specific PCR analysis (see below).
Sequence-specific PCR analysis
To validate insertion locus in HCD mutants, sequence-specific PCR assay was performed using primer pairs of HCD1-F and HCD1-R (5′-CCGTACATGCCGCACAGGAAGCCTAGGTAGCG-3′ and 5′-CCGAAGGCTGCACGTCCACTGACCTGGATTG-3′) for HCD1 and primer pairs of HCD3-F and HCD3-R (5′-CTCGACCATAAGGCGCCCGGCGTTTGAGTT-3′ and 5′-GCCTTCAACGACGCGAGGTTCAATACAGTGGAGC-3′) for HCD3. PCR analysis was carried out using 2× Taq Master Mix (Probegene Lte., Xuzhou, China) under the conditions of 95°C for 60 s; 95°C for 30 s, 64°C for 30 s, and 72°C for 80 s, those steps for 30 cycles; 72°C for 10 min. Nucleotide sequences flanking to the HCD1 and HCD3 loci are available in Supplementary Figs S2 & S3, respectively.
To examine the potential fusion transcript at the insertion locus, reverse transcription PCR assay was performed. Primers specific to Ble transcript (exon 2) Ble_E2_F2 (5′-GGTTCTCCCGGGACTTCGTGGAGGACGACTTC-3′) and flanking sequences at the exon 1 of Cre10.g426350 HCD1_R2 (5′-GTGGATCTCGAACTGCAAGGCGCACTAGGTGC-3′) for HCD1 and flanking sequences at the 5′-untranslated region (5′-UTR) of Cre13.g585600 HCD3_R2 (5′-TGGAGCTCGTTGGTGCAGGTAATGCCCTTGTG-3′) for HCD3. Nucleotide sequences of the fusion transcript with Ble in HCD1 and HCD3 are available in Supplementary Figs S4 & S5, respectively.
To test transcriptional levels of genes flanking to the insertion site, reverse transcription quantitative PCR (RT-qPCR) is performed in triplicate. Total RNA is extracted using TRIzol reagent (Takara Bio Inc., Tokyo, Japan). Total RNA of 500 ng is used for first strand cDNA synthesis using 500 ng total RNA with PrimeScript RT reagent kit with gDNA Eraser (Takara Bio Inc.) according to manufacturer’s protocol. The resulting cDNA is 10-fold diluted and subjected to quantitative PCR using SYBR Premix Ex Taq II kit (Takara Bio Inc.) on a Roche LightCycler 480 instrument (Roche) with the condition as follows: initial denaturing for 30 s at 95°C, followed by 40 cycles of 5 s at 95°C and 30 s at 58°C. Nucleotide sequences of gene-specific primers are as follows: for Cre10.g426350, HCD1L-fwd 5′-GCCAGCGAACCTGCGACAAG-3′ and HCD1L-rev 5′-GCAGCCAGGAGGGGAACAGC-3′; for Cre10.g426400, HCD1R-fwd 5′-GCGGCATGTACGGGCTCTTC-3′ and HCD1R-rev 5′-CCTCGGGCGTCAGCTTGTCC-3′; for Cre13.g585600, HCD3L-fwd 5′-AAGGTTGGCTTTTCGGGTCC-3′ and HCD3L-rev 5′-TGCTGTTGTTCGAGTTGGTGC-3′; for Cre13.g585651, HCD3R-fwd 5′-CTTAGCAGCGGTG GAAGAGC-3′ and HCD3R-rev 5′-GGCGGTGTCCGGGT CAATCC-3′. The TUB2 is used as control whose primers are TUB2-fwd 5′-CCCCCGCCTGCACTTCTTC-3′ and TUB2-rev 5′-GTCGGCGGCGCACATCAT-3′.
Transcriptome assembly analyses
Hi-Seq 2000 PE90 technique was used for transcriptomic analysis of HCD mutant cells. For this reason, cell samples were harvested and total RNA was extracted using Trizol reagent (Life Technology) according to manufacturer’s instructions. Resulting total RNA was subsequently treated with RNase-free DNaseI (Life Technology) and then column purified (RNeasy mini kit; Qiagen, Hilden, Germany). cDNA library construction for sequencing was performed in duplicate or triplicate by AIT Biotech (Singapore).
Approximately ~22 million good reads (90 nts in length) in each sample were subjected to Trimmomatic analysis (Bolger et al. 2014) in which 12 and 5 nucleotides at 5′ and 3′-ends (respectively) were trimmed. The resulting reads were subjected to TopHat analysis (Trapnell et al. 2009) to map reads based on reference genome of C. reinhardtii ver. 5.5 at Phytozome (https://phytozyme.jgi.goe.gov). Transcript abundances and differential transcription were obtained by using CuffLink and CuffDif analyses (Trapnell et al. 2009).
Meanwhile, the trimmed reads were also subjected to de novo assembly of transcripts using Trinity software aiming at finding novel transcripts at the insertion joint in HCD mutant strains when possible. Potential novel transcripts at the insertion locus were obtained by BLAST analysis (http://www.ncbi.nlm.nih.gov) of the Ble sequence and its flanking chromosomal sequences in HCD strains against the de novo assembled ESTs in respective mutant strains.
GO functional enrichment analysis
To investigate biological functions associated with the differentially transcribed genes in HCD1 and HCD3, gene ontology (GO) enrichment analysis is performed in the subset of the top 500 differentially transcribed (upregulated or down regulated) genes using binomial test. GO biological functions is considered when a total number of associated genes is 14 or greater. The function is enriched when the fold of enrichment is 2 or greater and p-value is less than 0.05.
Experimental replications and statistical analyses
All experiments including growth curve analysis, growth rate, maximum cell dry weight, and chlorophyll contents of wild type and HCD insertional mutant strains were based on the analysis of 3 independent biological repeats. Significant differences of sample measurements from that of wild type were calculated using t-test.
Original RNA sequencing (RNA-seq) data for transcriptome assembly using reference mapping-based and de novo approaches are available in SRA database (http://www.ncbi.nlm.nih.gov/sra) with an accession number SRP093432.
RESULTS
Enrichment of HCD mutant strains in continuous subcultures
To test whether quantitative genetic traits such as HCD would be enriched through continuous subcultures, we constructed over 1,000 individual insertional mutant strains generated through transformation of the PCR-amplified Ble expression cassette using the template pSP124S plasmid (Lumbreras et al. 1998) into wild type strain CC503 cells. Equal amount of individual insertional mutant strains was pooled and cultivated in triplicate in 100 mL HS medium (Sueoka 1960) with 70–90 rpm shaking at room temperature under the continuous illumination of 50 μmol photon m−2 s−1, bubbling with ~2% CO2 (see Materials and Methods). Under this condition, the starting culture (~0.15 OD750) of the mixed mutant cells reached to ~0.4, ~0.95, ~1.35, and ~1.45 OD at day 1–4, respectively (Fig. 1A). Cells at day 3 entered the onset of stationary-phase and hence were used as inoculum to start the cycles of continuous subcultures (Fig. 1B). Subcultures were continued for 30 cycles.
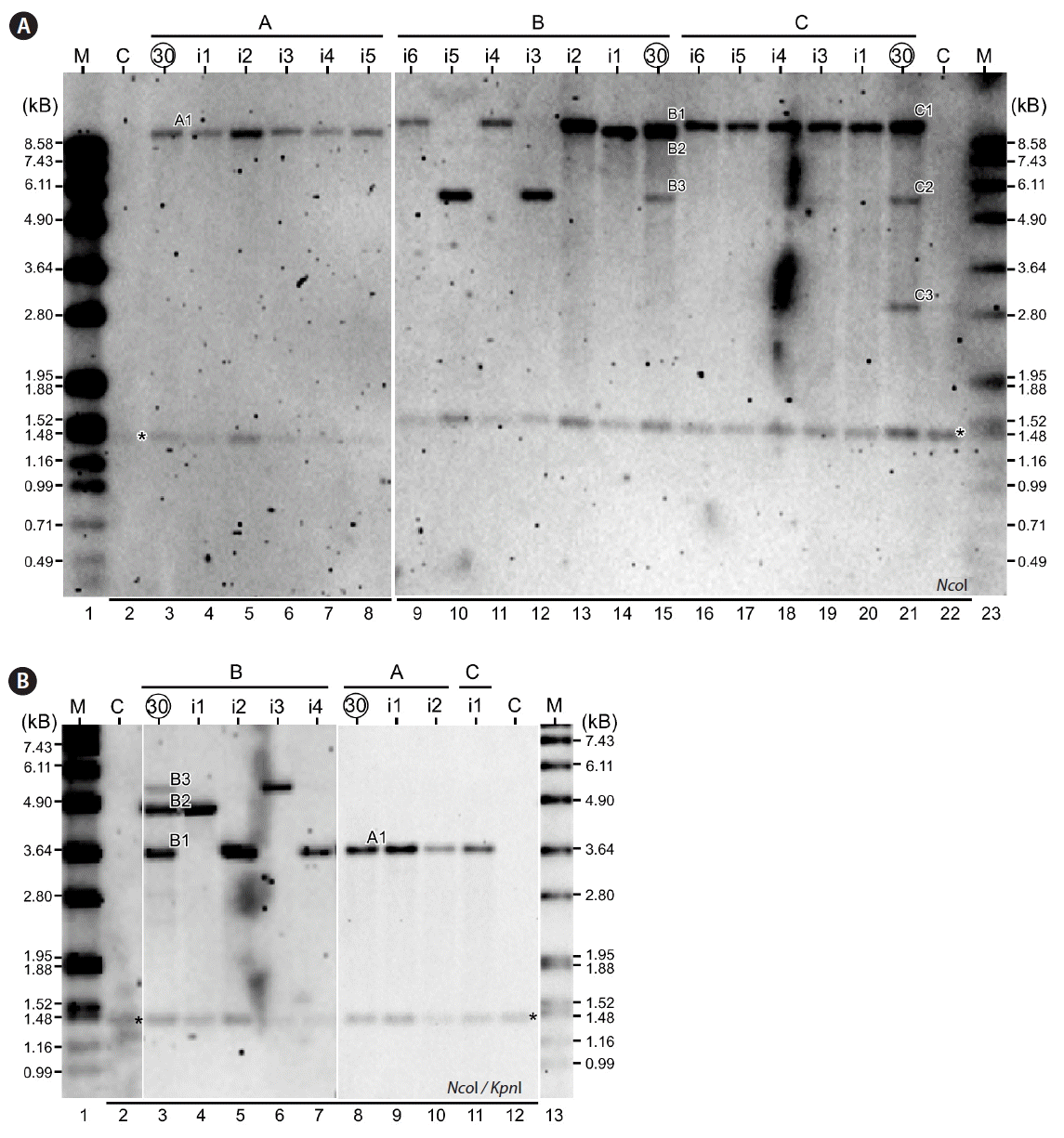
Mutant strains exhibiting HCD phenotypes would be expected to dominate the cultures containing over a thousand mutant populations after multiple rounds of subcultures. To investigate whether HCD mutant strains were gradually enriched in the continuous subcultures, RFLP pattern of the Ble expression cassette in NcoI digested genomic DNA derived from initial culture and subcultures after 10, 20, and 30 cycles were analyzed (Fig. 1C). A complex RFLP pattern was apparent in the initial culture containing the pool of a thousand insertional mutant strains (Fig. 1C, lane 3). We found that the complexity of RFLP patterns decreased upon increase of subculture cycles, suggesting that gradual enrichment of the HCD mutant strains occurred during continuous subcultures. In three independently repeated experiments A–C, 1–3 fragments (i.e., fragments A1, B1–3, and C1–3) in RFLP analysis were found after 30 cycles of subcultures. Strains bearing one of these fragments would be candidates of HCD strains.
Isolation of enriched HCD mutant strains from cultures after 30 cycles of subcultures
To isolate strains bearing one of the dominant RFLP fragments, cultures after 30 cycles of subcultures were plated out on solid HS plates for single colonies. Five to six randomly selected colonies from each repeated experiment were subjected to RFLP analysis using the NcoI digested genomic DNA. All 5 isolates from experiment A showed to have the fragment A1 (Fig. 2A, lanes 4–8). In experiment B, isolate 2, 4, and 6 possessed the fragment B1 (Fig. 2A, lanes 9, 11, and 13), while isolate 3 and 5 contained the fragment B3 (Fig. 2A, lanes 10 and 12). One of the isolates in experiment B showed to have the fragment B2 (Fig. 2A, lane 14). In experiment C, all 5 isolates showed to have the fragment C1 (Fig. 2A, lanes 16–20). None of the isolates tested was found to possess the fragment C2 or C3.
To confirm the isolates contained the enriched fragments after 30 cycles of continuous subcultures based on the RFLP analysis using NcoI single digested genomic DNA, we performed the RFLP analysis using NcoI and KpnI double digested genomic DNA. Although the sizes of enriched fragments altered in double digestion, relationship between isolates and its RFLP patterns remained identical to that of single digestion (Fig. 2B), supporting the RFLP analysis based on single NcoI digestion (see Fig. 2A). Hence, we designated the mutant strains containing the fragments B1 (isolates A1_i1-i5; B1_i2, i4, and i6; and C1_i1 and i3–i6), B2 (isolate B2_i1), and B3 (B3_i3 and i5) as HCD1, HCD2, and HCD3, respectively.
HCD mutant strains exhibit high maximum cell-culture density phenotypes
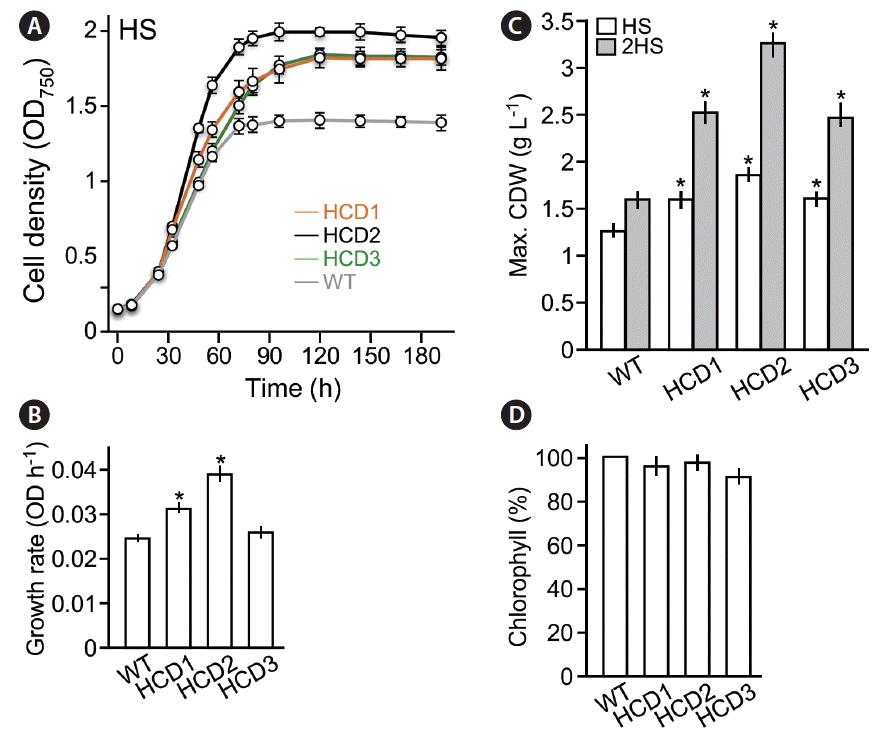
To investigate the maximum cell density of the HCD mutant strains, growth curves analyses were performed under photoautotrophic growth conditions (HS medium) with continuous illumination at 50 μmol photon m−2 s−1 (Fig. 3A). We found that the growth rates (i.e., average growth rate at the log-phase from 24 to 56 h based on triplicate) of HCD1 and HCD2 but not HCD3 were significantly higher than that of wild type (p < 0.05) (Fig. 3B). On the other hand, the maximum cell densities of HCD1, HCD2, and HCD3 in HS medium were 22–40% higher than that of wild type (p < 0.05) (Fig. 3C, open bar). When cells were cultivated in 2-times concentrated HS medium (2HS medium), the maximum cell density of the HCD mutant strains were 50–100% higher than that of wild type (p < 0.05) (Fig. 3C, grey bar).
To investigate the level of chlorophyll contents in HCD mutant strains, we determined the ratio of chlorophyll and cell dry weight. Percent of chlorophyll contents in cell dry weight of HCD mutant strains were not significantly lower than that of wild type (Fig. 3D), suggesting that the HCD phenotypes are unrelated to that of truncated light-harvesting antenna TLA mutants (Polle et al. 2003). This result is consistent with the notion that biomass yield of TLA mutant strains increases under growth conditions with high illumination such as >500 μmol photon m−2 s−1 (Polle et al. 2003) but not low illumination (e.g., ~50 μmol photon m−2 s−1).
Identification of insertion loci in HCD mutant strains using inverse-PCR methodology
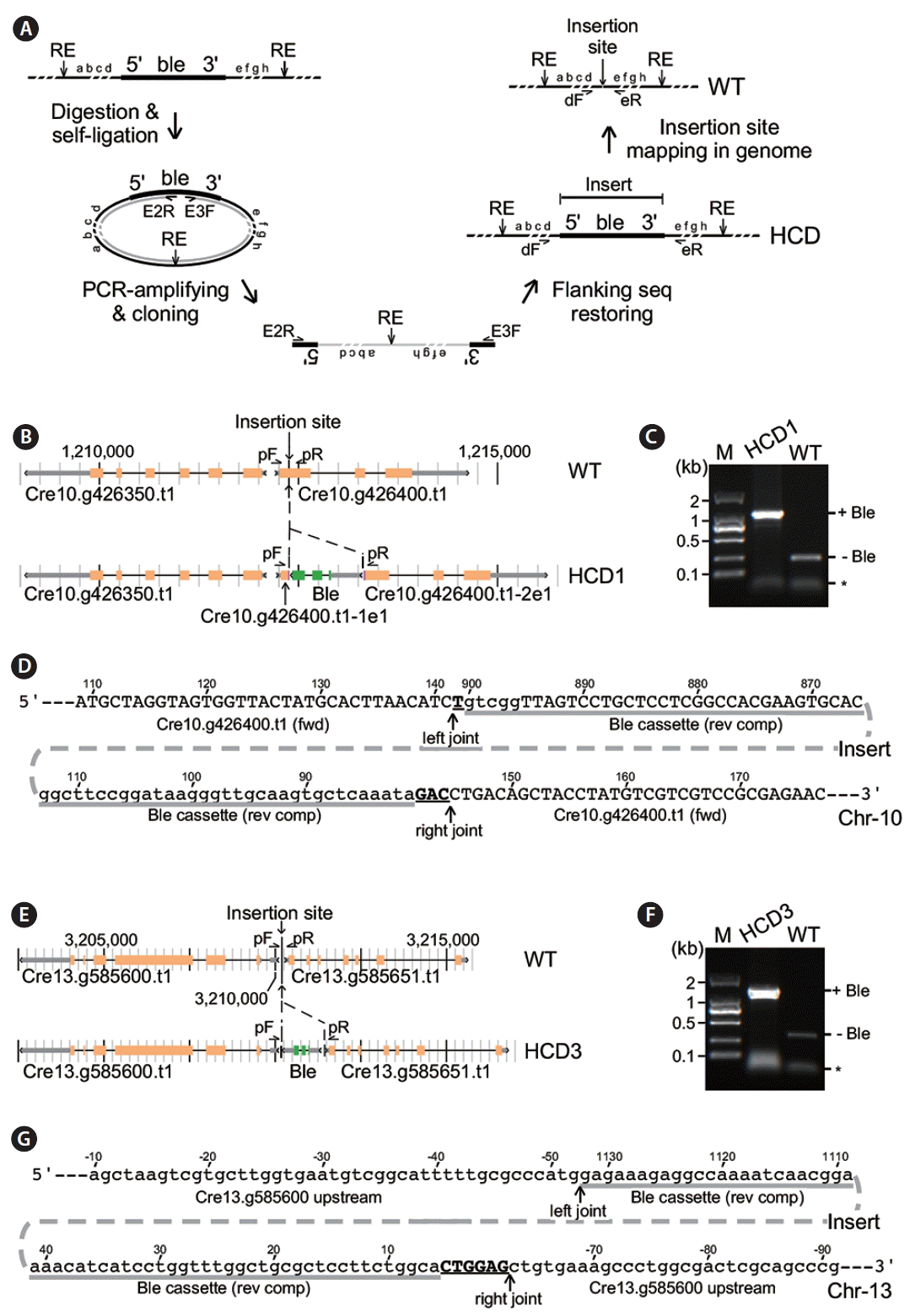
Given that HCD1, HCD2, and HCD3 exhibited a single fragment in RFLP analysis of both single and double digestions (see Fig. 2), we assumed that only a single copy of the Ble expression cassette was inserted into the genome of individual HCD mutant strains. Hence, we mapped the insertion locus in HCD mutant strains using inverse-PCR analysis (Yoshioka et al. 2004). For this reason, genomic DNA of HCD mutant cells was subjected to single digestions of various restriction enzymes including but not limited to NheI, NdeI, and PstI that had no recognition site in the Ble cassette insert (Fig. 4A). The resulting DNA was then self-ligated with T4 DNA ligase and followed by PCR amplification using sequence-specific primers E2R (Ble exon 2 reverse) and E3F (Ble exon 3 forward) (see Materials and Methods). PCR fragments were successfully amplified in HCD1 and HCD3 but not HCD2 after self-ligation.
Sequencing analysis of the inverse-PCR fragments generated from HCD1 suggested that the Ble cassette was inserted in the first exon of Cre10.g426400 (Fig. 4B). We applied PCR analysis using sequence-specific primers flanking to the Ble insert to confirm its insertion locus in HCD1. We found that the identical pair of primers gave rise to a fragment of ~1.1 kb in size using HCD1 genomic DNA as template, which was ~850 bp longer than the fragment of ~250 bp in size using wild type CC503 genomic DNA as template (Fig. 4C). This result confirmed that the Ble cassette was inserted into the first exon of Cre10.g426400, splitting it into two parts: Cre10.g426400-1e1 (or transcript containing the first part of exon 1) and Cre10.g426400-2e1 (or transcript containing the second part of exon 1). Sequencing analysis of the PCR fragment encompassing the HCD1 locus indicated that the Ble cassette was inserted in a reverse orientation into the first exon of Cre10.g426400 at positions of 141 and 145 nt with a 3-nt deletion in Cre10.g426400 (Fig. 4D). The insert contained Ble cassette sequences from positions 81 to 900 nt missing the entire 3′-UTR. Meanwhile, we found that a single nucleotide and trinucleotide were inserted in the left and right joints flanking the Ble insert, respectively.
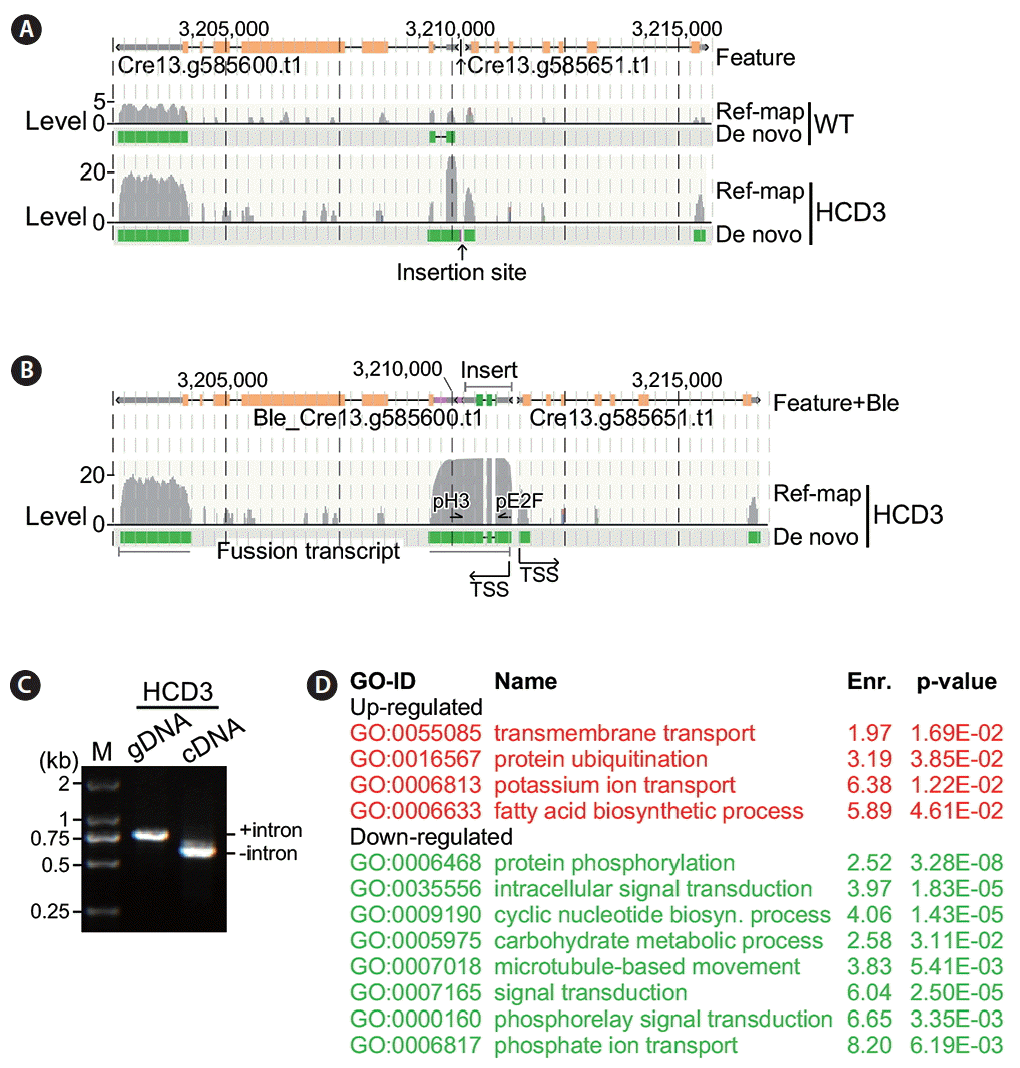
Similarly, inverse-PCR analysis of HCD3 indicated that the Ble cassette was inserted into the intergenic region between Cre13.g585600 and Cre13.g585651 (Fig. 4E). PCR analysis using sequence-specific primers showed that fragments of ~1.4 kb and 300 bp in size were amplified in HCD3 and wild type (respectively), indicating a fragment of ~1.1 kb in size was present at the insertion locus of HCD3 (Fig. 4F). Sequencing analysis of the PCR fragment encompassing the HCD3 locus indicated that most of the Ble cassette sequences were inserted into the intergenic region at positions from −52 and −63 nt with a deletion of 10 nt based on the position of Cre13.g585600 (Fig. 4G). The Ble cassette sequences from positions 6 to 1,132 nt (the whole Ble cassette was 1.152 kb in length out of the full-length PCR fragment of 1.174 kb) were found in the insertion locus of HCD3. The left joint showed no nucleotide addition. On the other hand, a hexanucleotide were found in the right joint. These results indicated that part of the terminal sequences in the Ble cassette could be lose during insertion into genome. In addition, single and short stretches of nucleotides such as trinucleotide and hexanucleotide could be added during random insertion.
Transcription enhancement of the disrupted partial transcript and the Ble-fusion transcript at the HCD1 locus
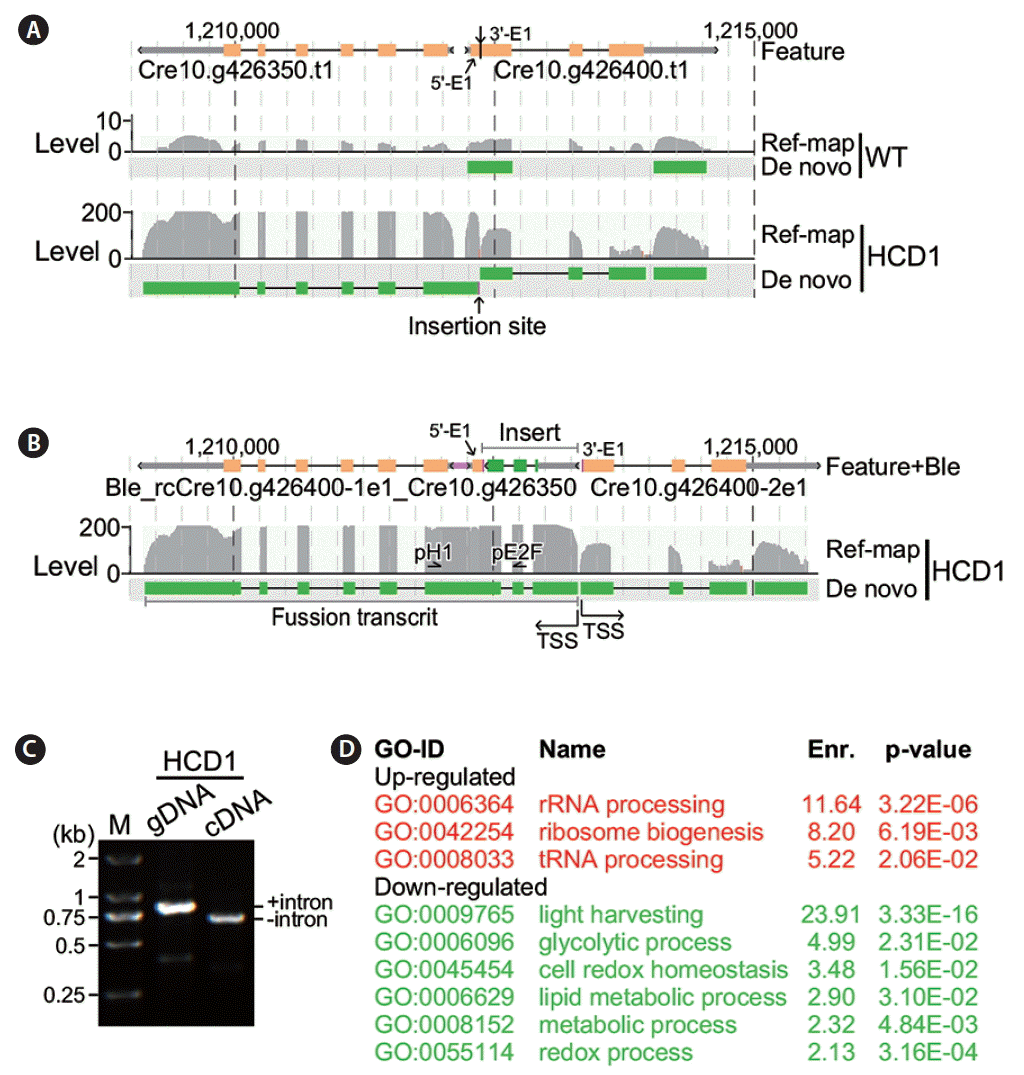
To investigate whether the transcription of genes flanking to the insert at the HCD1 locus was disrupted in the mutant strain, we performed transcriptional profiling analysis of the HCD1 and wild type CC503 strain (see Materials and Methods). In this analysis, RNA samples was subjected to library construction for sequencing using illumine HiSeq technology in triplicate (see Materials and Methods). Transcriptomes was assembled using reference-based methodology (Trapnell et al. 2009) (Supplementary Table S1). In wild type cells, transcription levels of Cre10.g426400 at the HCD1 locus was 6.26 fragments per kilobase of exon per million fragments mapped (FPKM), based on the average of three repeated measurements (all measurements of transcriptional levels were derived from the average of three repeats) (Fig. 5A, upper track). We found that the transcription level of Cre10.g426400 was 39.66 FPKM in HCD1 cells, which was 6.33-fold higher than that of wild type cells (p = 2.4e-04) (Fig. 5A, lower track). Meanwhile, we found that the transcription level of Cre10.g426350, upstream of Cre10.g426400, was 3.89 FPKM in wild type. The level of Cre10.g426350 was increased to 170.89 FPKM in HCD1, indicating that Ble cassette insertion induced the transcription of Cre10.g426350 by 44-fold.
We hypothesized the promoter P-RBCS2 in the Ble cassette was responsible for the bi-directional activation of transcription at the HCD1 locus. To test this possibility, we performed the de novo assembly (Grabherr et al. 2011) of transcriptomes of HCD1 mutant. Analysis of the de novo assembled transcripts containing the HCD1 flanking gene sequences indicated that the transcription start site (TSS) of Cre10.g426400-2e1 (i.e., transcript containing the second part of exon 1) was located at boundary of the right joint. On the other hand, reverse-complementary sequences of Cre10.g426400-1e1 was found in a fusion transcript containing Ble, reverse-complementary (rc) Cre10.g426400-1e1, and Cre10.g426350 sequences (Fig. 5B). To investigate the presence of the fusion transcript containing Ble, rcCre10.g426400-1e1, and Cre10.g426350 sequences, we performed PCR analysis using sequence-specific primers located at Ble exon 2 and Cre10.g426350 exon 1 using either HCD1 genomic DNA or cDNA as template. In this case, the two primers would amplify a fragment encompassing the intron 2 of 145 bp in size located between Ble exon 2 and Cre10.g426350 exon 1 in the HCD1 genomic DNA but not cDNA. The result showed while a fragment of ~850 bp in size was amplified using genomic DNA as template, a fragment of ~700 bp in size was amplified using cDNA template, supporting the existence of the fusion transcript containing Ble, rcCre10.g426400-1e1, and Cre10.g426350 sequences (Fig. 5C).
Subsequently, we performed functional enrichment analysis in top 500 differentially transcribed genes in HCD1 (see Materials and Methods). Analysis indicated that functions related with ribosomal biogenesis (GO:0006364, GO:0042254, and GO:0008033) were enriched in the top 500 significantly upregulated genes in HCD1 (Fig. 5D). There were six GO functions (GO:0009765, GO:0006096, GO:0045454, GO:0006629, GO:0008152, and GO:0055114) were found to be enriched in the top 500 significantly downregulated genes in HCD1, two of which were related to the putative thioredoxin function of the disrupted Cre10.g426400.
Intergenic insertion induces transcription of flanking genes in HCD3 locus
To investigate alteration of transcriptions at the HCD3 insertion site, we performed reference-based and de novo transcriptome analyses of HCD3 mutant strain and found that the transcriptional levels of Cre13.g585600 and Cre13.g585651 were increased by 3.31-fold (8.57 FPKM vs. 3.31 FPKM, p = 2.4e-04) and 2.21-fold (2.66 FPKM vs. 1.21 FPKM, p = 4.9e-02) in HCD3 cells compared to that of wild type (Fig. 6A). Analysis of de novo transcripts containing the flanking gene sequences indicated that the TSS of Cre13.g585651 was not altered in HCD3 compared to that of wile type (Fig. 6B). On the other hand, Cre13.g585600 transcript was found to be fused with Ble sequences, that was inserted at the upstream of Cre13.g585600 with the same orientation. To confirm the presence of the fusion transcript between Ble and Cre13.g585600, we performed PCR analysis using sequence-specific primers located at Ble exon 2 and Cre13.g585600 exon 1 using either HCD3 genomic DNA or cDNA as template. It was clear that the intron sequence was excised in cDNA or transcript but not in genomic DNA (Fig. 6C).
Functional enrichment analysis indicated that, in HCD3, four GO functions (GO:0055085, GO:0016567, GO:0006813, GO:0006633) were enriched in the top 500 significantly upregulated genes (Fig. 6D). Lon protease was proposed to be linked to the proteasome function (Erjavec et al. 2013). While Cre13.g426400 encoded a putative Lon protease, upregulation of protein ubiquitination function is consistent with its upregulation in HCD3. On the other hand, eight GO functions (GO:0006468, GO:0035556, GO:0009190, GO:0005975, GO:0007018, GO:0007165, GO:0000160, and GO:0006817) were enriched in the top 500 significantly downregulated genes in HCD3.
Phenotypes associated with HCD1 and HCD3 mutants
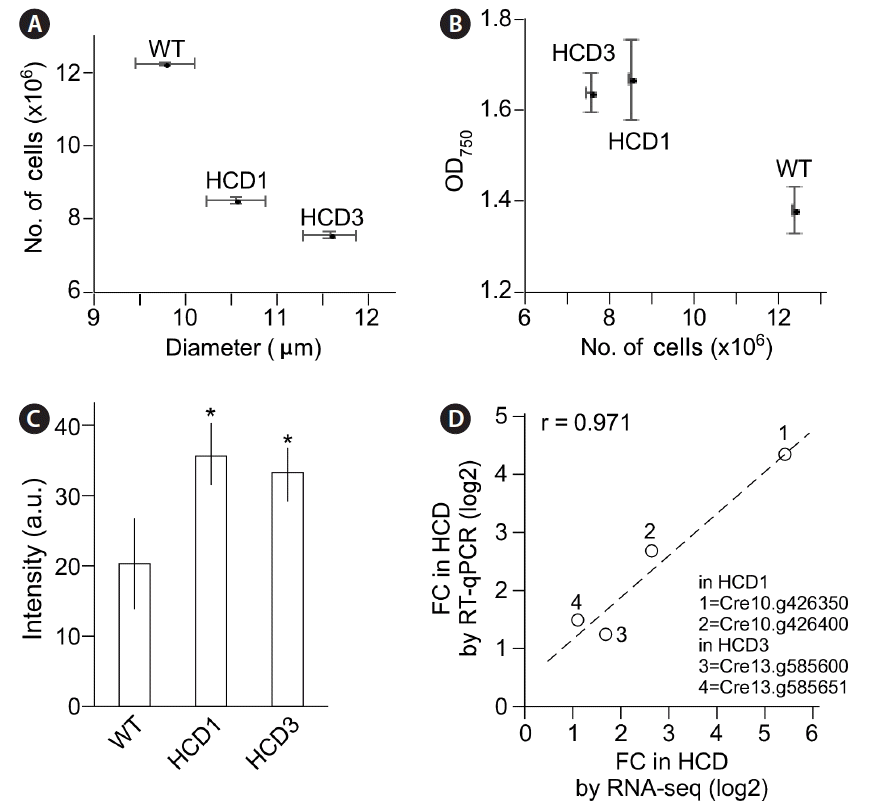
We found that, at the onset of stationary phase (e.g., 96 h after growth), the median diameter of HCD1 and HCD3 cells was 10.6 and 11.6 μm, which was 7 and 17% bigger than that of wild type (9.9 μm) (p = 0.0012 and p = 8.24e-05, respectively) (Fig. 7A & B). To test whether the size of mitochondria in HCD1 mutant cells altered as well, cells were subjected to mitotracker fluorescence dye staining (see Materials and Methods). The mitochondrial sizes were approximated by the average fluorescence signals. To this end, we found that the average fluorescence signals in HCD1 and HCD3 were 53.7 and 33.1 (arbitrary unit), which was 1.77-fold and 1.64-fold higher than 20.2 a.u. of wild type (p = 2e-06 and p = 1e-05, respectively) (Fig. 7C).
Transcription levels of genes at the insertion site in HCD1 or HCD3 were found to be altered in RNA-seq analysis (see Figs 5A & 6A). To validate the change, RT-qPCR was performed. We found that the fold changes of transcription levels of at the insertion site in HCD1 or HCD3 in RT-qPCR and RNA-seq analyses were well correlated with a correlation coefficient of 0.971 (Fig. 7D).
DISCUSSION
Increasing microalgal maximum cell-culture density is one of the ways for improving its biomass productivity. Early works have shown that C. reinhardtii cells with reduced size of light-harvesting antenna or lowered chlorophyll content exhibit increased cell-culture density under high-illumination growth conditions (Polle et al. 2003). In this study, we show that C. reinhardtii mutant strains without lowering the chlorophyll content display the HCD phenotype under low-illumination condition (see Fig. 3).
It has been proposed that only a small fraction of the light harvested by algal cells with large-sized antenna is converted to chemical energy under high-illumination (Polle et al. 2003). Hence, small-sized antenna allows more cells to access light and thus increases the efficiency of light usage. In this study, we show that in HCD1, the insert disrupted the intragenic sequences of Cre10.g426400, leading to the increase of transcription levels of the truncated Cre10.g426400-2e1 transcript and the fusion transcript containing Ble, rcCre10.g426400-1e1, and Cre10.g426350 (see Fig. 5). While the function of Cre10.g426350 is unknown, Cre10.g426400 encodes a putative thioredoxin. Transcription profile analysis indicates that ribosomal biogenesis functions (GO:0006364, GO:0042254, and GO:0008033) are enriched in the top significantly upregulated genes in HCD1, consistent with its high biomass yield. Additionally, redox processes (GO:0045454 and GO:0055114) are enriched in the most downregulated genes, in agreement with the disruption of the putative thioredoxin gene Cre10.g426400.
In HCD3, the Ble cassette was inserted at the intergenic region between Cre13.g585600 and Cre13.g585651, leading to the transcription level increase of Cre13.g585651 and a fusion transcript containing Ble and Cre13.g585600 (see Fig. 6). Though the function of Cre13.g585600 is unknown, Cre13.g585651 encoded a putative mitochondrial ATP-dependent Lon protease. Products of both Cre10.g426400 in HCD1 and Cre13.g585651 in HCD3 are potentially linked to mitochondrial function (Kang et al. 2002, Folda et al. 2016). We show that both HCD1 and HCD3 exhibit elevated mitotracker fluorescence staining, suggesting a link to mitochondrial function in increased cell density phenotype. Hence, we propose that abnormality of mitochondrial function in HCD1 and HCD3 play a role in increased cell-culture density under low-illumination condition.
In this study, we show that mutant strains exhibiting quantitative traits of HCD are enriched from the pool of the randomly mutated strains through continuous subcultures. Analysis of clonal HCD isolates confirms their high biomass yield under low-illumination growth conditions. Molecular analysis indicates that transcription of flanking genes is upregulated by the intragenic and intergenic insertions in HCD1 and HCD3, respectively. In both cases, transcriptionally enhanced genes flanking to the insertion site with or without intact sequences are potentially linked to the mitochondrial functions (Kang et al. 2002, Folda et al. 2016). We propose that by using continuous subcultures methodology, more HCD strains can be enriched from a large pool of insertional mutant strains. Analysis of novel HCD strains will allow gaining insight into mechanisms for regulation of cell-culture density under various illumination conditions in C. reinhardtii and other green algal species.