Abbreviations
CA
cellulose acetate
CO1
mitochondrial cytochrome c oxidase subunit 1
ddPCR
droplet digital polymerase chain reaction
eDNA
environmental DNA
HABs
harmful algae blooms
ITS
internal transcribed spacer
ML
maximum likelihood
PCR
polymerase chain reaction
qPCR
quantitative real-time polymerase chain reaction
rbcL
chloroplast rubisco (ribulose-1,5-bisphosphate carboxylase/oxygenase) large subunit
SSU
small subunit of rDNA
INTRODUCTION
Nationwide monitoring of harmful algal blooms (HABs) has been conducted either monthly or bimonthly in Korea in all seasons except winter since early 2000 as many fish and shellfish farms are located in shallow waters (http://www.nifs.go.kr/). To date, this monitoring data have contributed to our understanding of the impacts and dynamics of HABs (Jang and Jeong 2020, Lee et al. 2020, 2021a, You et al. 2020, Eom et al. 2021) and, fortunately, were not reported for ichthyotoxic or fish-killing blooms of Chattonella species.
The Chattonella genus was first described by Biecheler (1936) with the type species C. subsalsa. The genus comprises seven species and two varieties (Guiry and Guiry 2021). The cells of the genus Chattonella are pyriform in shape, with two laterally placed flagella and tightly packed plastids along the cell periphery, which can be anteriorly rounded and posteriorly angulated because they lack a rigid cell wall (Klöepper et al. 2013). Both C. marina and C. marina var. ovata were described based on morphological differences observed in the overall size and shape of the cell, which were visible to light microscopy (Demura et al. 2009). Two species have subsequently been shown to have significant sequence similarities for multiple phylogenetic markers (internal transcribed spacer [ITS], large ribosomal subunit, rbcL, mitochondrial cytochrome c oxidase subunit 1 [CO1]), which allowed them to be classified as belonging to a species complex (Demura et al. 2009, Klöepper et al. 2013).
Chattonella blooms have been reported in Asia (Kim et al. 2007, Imai and Yamaguchi 2012, Wang et al. 2017, Yamaguchi et al. 2018), Oceania (Munday and Hallegraeff 1998, Marshall and Hallegraeff 1999), North America (Lewitus et al. 2008, García-Mendoza et al. 2018), and Europe (Stacca et al. 2016, Satta et al. 2017, Zingone et al. 2021). The ichthyotoxic mechanism by Chattonella blooms is thought to be caused by the toxicity of polyunsaturated fatty acids that increase synergistically in the gills during the production of reactive oxygen species, such as superoxide anions and hydrogen peroxide released from Chattonella (Marshall et al. 2003, Dorantes-Aranda et al. 2015). The Chattonella cells become trapped within the fish mucus, which can lead to gill damage and eventual suffocation (Shen et al. 2011).
As a unicellular photosynthetic flagellate, natural populations of Chattonella exhibit a diel vertical migration behavior, whereby individuals cells swim up to the surface layers to photosynthesize during the daytime and sink downward at night to take up nutrients from the benthic layers (Watanabe et al. 1991, Shikata et al. 2019, Qiu et al. 2020). Chattonella species have been described as mixotrophs that feed on various heterotrophic and autotrophic bacteria and cyanobacteria (Jeong 2011). In addition, the maximum growth rate was found to be slightly higher in C. marina than in C. marina var. ovata, although thermal tolerance was slightly greater in C. marina var. ovata than in C. marina (Lim et al. 2020). Some of Chattonella species have high physiological plasticity, enabling them to adapt successfully to climate change and colonize diverse habitats (Lim et al. 2020, Vidyarathna et al. 2020).
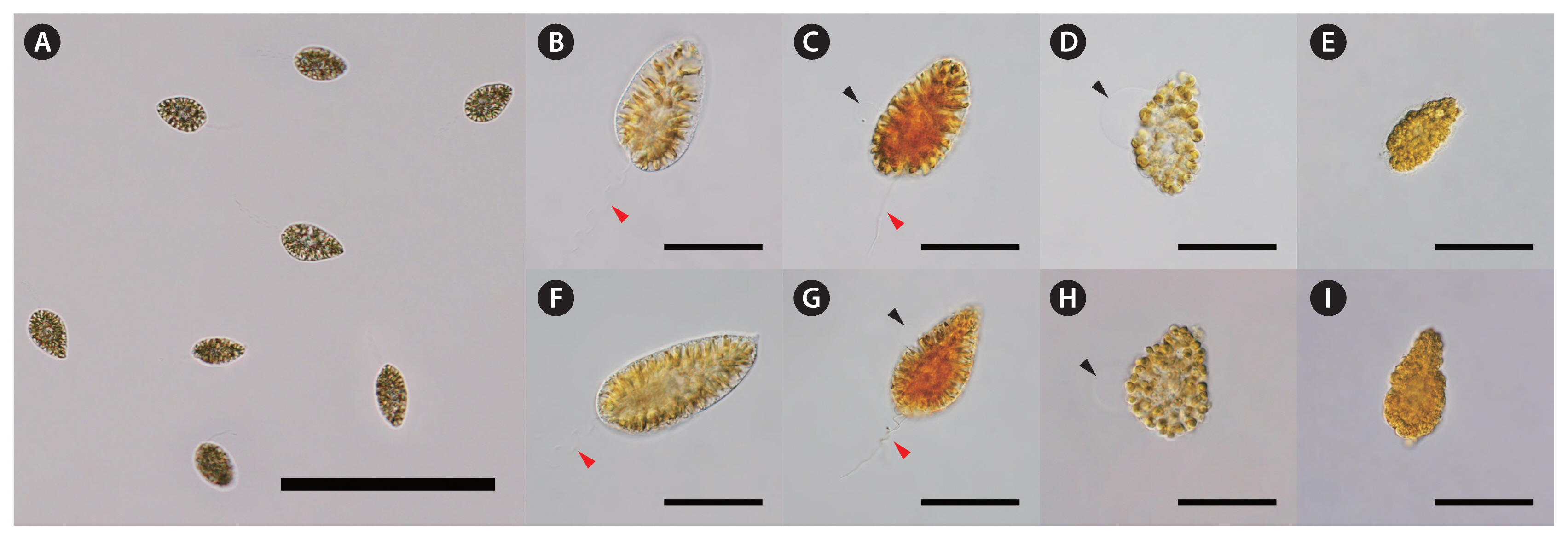
Identifying Chattonella cells at the species level under a microscope is difficult because of their morphological plasticity and fragile cell nature (Coyne et al. 2005). Furthermore, Chattonella cells disintegrate with most fixatives and display peculiar morphological features (Klöepper et al. 2013). The size of the cell vacuoles may be the only characteristic that allows discrimination between C. marina and C. marina var. ovata, but it is not always easy to establish size boundaries (Demura et al. 2009). Cell counting among Chattonella is not possible at the species level, only genus, or species complexes at best. Additionally, microscopic observation, which is helpful for preliminary identification, can be altered by mechanical damage and preservation artifacts.
Several studies have explored the application of environmental DNA (eDNA)-based tools such as quantitative real-time polymerase chain reaction (qPCR) in conducting quantitative assessments of marine plankton. qPCR-based eDNA analyses targeted toward individual species showed a higher level of detection efficiency and had greater accuracy at low abundance (Wood et al. 2019). However, discrepancies between the qPCR and microscope counts were observed during HAB monitoring (Murray et al. 2019, Lee et al. 2021b, Ok et al. 2021). This may be caused by several factors regarding polymerase chain reaction (PCR)-based quantification, such as the choice of the most appropriate genetic marker and the efficiency of the PCR amplification of the target DNA (Raeymaekers 2000, Jin et al. 2020).
We hypothesized that the copy number of the ITS region correlates with Chattonella cell abundance so that the ITS copies in each cell can be applied to the absolute quantification. This method assumes that gene copy number is relatively constant within a species and differs by no more than two-fold over the cell cycle. A droplet digital polymerase chain reaction (ddPCR) assay allows the cell abundance of species or taxon in an eDNA sample to be detected using a short DNA sequence specifically designed for a particular species or taxonomic group (Lee et al. 2017). We present an integrated approach for the species-specific quantification of dinoflagellate Alexandrium species, which applies a combination of light microscopy coupled with a ddPCR assay with excellent accuracy and sensitivity (Lee et al. 2020). This advanced protocol based on the ddPCR technique, which was proposed to quantify species’ cell abundance, was particularly suited to analyzing large numbers of samples with a higher level of accuracy and resolution than microscopy analyses. This technique is essential for HAB monitoring, where thousands of samples must be analyzed quickly and reliably, even at low cell abundance.
Since microscopic observation alone cannot routinely monitor the Chattonella taxa, new approaches to quantification at the species or group level need to be developed and implemented to assist in timely diagnosis and appropriate intervention of Chattonella blooms. Detecting species at low abundance level is critical for managing HAB events, as early detection is crucial. Therefore, this study aimed to design specific detection primers and develop a ddPCR assay for quantifying C. marine and C. marina var. ovata that are associated with fish-killing event in coastal waters.
MATERIALS AND METHODS
Sample preparation
The C. marina culture (CM211006MP1) was isolated from Mokpo in October 2021, while C. marina var. ovata (COKP9909) was acquired from cultures isolated from Gyeokpo in September 1999 (Fig. 1). Chattonella cells were incubated in f/2 medium at 24°C and 150 μmol photons m−2 s−1 with a 14 : 10 h light-dark cycle and were subcultured at weekly intervals. Cells used for the experiment were collected during the exponential growth phase.
Phylogenetic analysis
For the sequence analysis and conventional PCR, the genomic DNA of two species was extracted from culture strains using a DNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA). In order to explore the phylogenetic placement of Chattonella, the gDNA was amplified using primer sets targeting a partial region of the ITS ribosomal DNA (ITS-rDNA), chloroplast rbcL, and mitochondria CO1 genes (Table 1). The amplification process consisted of an initial denaturation at 95°C for 10 min, followed by 32 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 30 s, with a final extension at 72°C for 5 min in a T100 Thermal Cycler (Bio-Rad Lab Inc., Hercules, CA, USA). The PCR reaction mixture had a total volume of 20 μL, consisting of 10 μL of 2× prime Taq premix (Genet Bio, Nonsan, Korea), 1 μL of 10 pmol of each primer (forward and reverse), and 80 ng DNA templates. The PCR products, purified using an Expin PCR SV Kit (GeneAll, Seoul, Korea) were sequenced using an ABI 3730XL DNA analyzer (Applied Biosystems, Foster City, CA, USA).
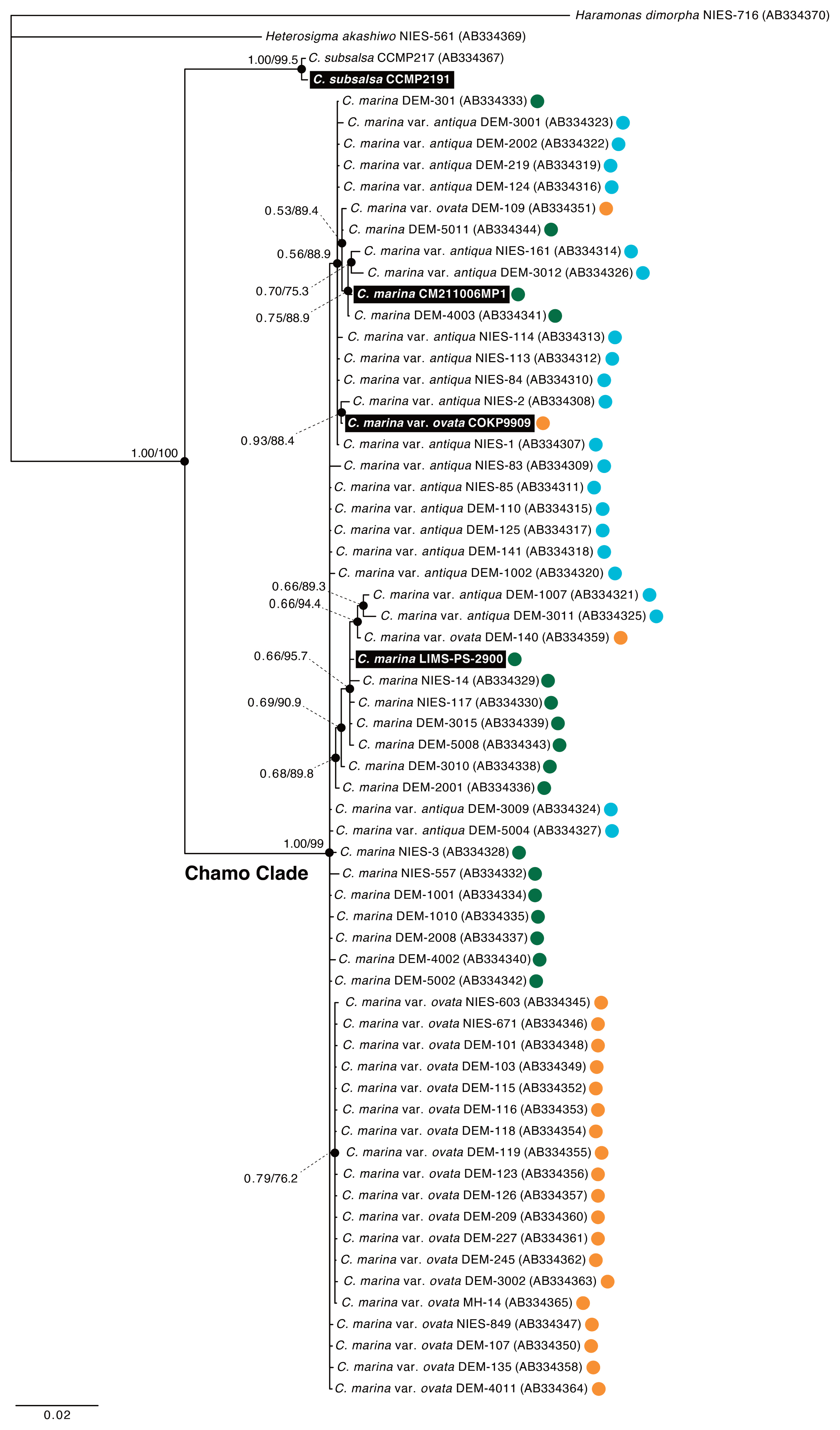
The sequences were aligned including sequences obtained from NCBI GenBank (Demura et al. 2009), as a trimming of the non-alignment sites, collected approximately 620 bp of the ITS-rDNA, 550 bp of rbcL, and 470 bp of CO1 gene. All three sequences were concatenated using MEGA X v10.2.4 (Stecher et al. 2020). Phylogenetic trees were constructed using the maximum likelihood (ML) and Bayesian inference methods. ML analysis was conducted using IQ-TREE v2.0.3, with the best substitution models for each gene with rapid bootstrapping and 1,000 replicates using a partitioned analysis (Chernomor et al. 2016). Bayesian analysis was conducted using MrBayes v.3.2.7 (Ronquist et al. 2012) with the general time-reversible model, along with additional invariant sites and a gamma distribution of rates across sites (GTR + G + I) in order to select the best available model for the data. The Markov chain Monte Carlo simulation was performed for all sequence regions until the average standard deviation of split frequencies reached <0.01. The consensus tree which added tree statistics as annotations was made by a convergence of trees sampled every 1,000 generations, and it was visualized using FigTree v1.4.4. (Rambaut 2018).
Specific primer design
The C. marina complex-specific primers were designed based on ITS sequences between 18S small subunit (SSU) and 28S large subunit rDNA that possess highly conserved sequences and are widely used in molecular phylogenetic studies. The copy number of tandem repeat units varied between species (Warmerdam and Wolthuis 2019). The specificity of the primers was validated in silico using the NCBI Primer-BLAST tool (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). The conventional PCR was used to be in vitro verified their specificity by gDNA extracted from five strains of raphidophytes containing Chattonella species, 12 strains of dinoflagellates, and one diatom, using the DNeasy Plant Mini Kit (Qiagen). The PCR reaction was performed in the same process detailed above, except that there were 40 cycles of amplification and at 59°C of annealing with 1 μL of 2 pmol of each primer.
Assay and validation of ddPCR
We filtered the culture sample containing known cell counts through a 0.45 μm cellulose acetate (CA) filter to determine the ITS copy number of species. We also collected various environmental samples from different locations, including Wando (34°19′17″ N, 126°44′54″ E), Yeosu (34°37′48″ N, 127°37′42″ E), Busan (35°4′37″ N, 129°4′57″ E), and Uljin (37°4′19″ N, 129°24′50″ E) during September 2022. We filtered them by adding known cell counts of C. marina and C. marina var. ovata in the same way to confirm inter-sample variability and the consistency of the ddPCR assay.
DNA for ddPCR was extracted from cultures and environmental samples through a CA filter using a previously reported two-step method (Lee et al. 2020). A whole CA filter was transferred to a 5 mL microfuge tube. Cells were pulverized by bead-beating (FastPrep-24; MP Biomedicals Inc., Santa Ana, CA, USA) with 2 mm zirconia beads (Watson Co., Tokyo, Japan) and lysed by incubating in 2 mL of lysis buffer (25 mM NaOH and 2 mM EDTA) at 95°C for 30 min. Next, after 2 mL of 40 mM Tris-HCl (pH 5.5) was added to neutralize the sample DNA, 5 μL of the DNA-containing supernatant was used as the template DNA of ddPCR.
Following the manufacturer’s instructions, a QX200 ddPCR system was used to determine the copy number per cell of each species of the C. marina complex (Bio-Rad Lab Inc.). The ddPCR reaction mixture comprised a total volume of 20 μL, containing 10 μL of EvaGreen Supermix (Bio-Rad Laboratories Inc., Munich, Germany), 1 μL each of 2 pmol of forward and reverse primers, 5 μL of sample DNA, and sterile water to make it up to the final volume. Each reaction mixture was partitioned into approximately 20,000 droplets using a Bio-Rad QX200 Droplet Generator (Bio-Rad Lab Inc.), and amplified using a T100 Thermal Cycler (Bio-Rad Lab Inc.). After the PCR amplification, each droplet in the sample was read using a droplet reader (QX200 Droplet Reader; Bio-Rad Lab Inc.) to determine as positive or negative and amplitude according to a fluorescence threshold signal level. QuantaSoft version 1.7.4 (Bio-Rad Lab Inc.) was used to calculate the number of target DNA copies from the fraction of positive end-point reactions using Poisson statistics. To ensure optimal detection, a minimum number of accepted droplets were required more than 10,000, and the saturation samples that the number of positive droplets occupied above 63% of accepted droplets were excluded (Pinheiro et al. 2012). To evenly eliminate false-positive and false-negative signals, the threshold assorted negative and positive droplets was established at the point of amplitudes (ca. 5,500) having no droplets in the no template controls.
To examine the reproducibility of the ddPCR assay, we analyzed the relationship between the copies and cell counts (spiked with various known cell counts) and the copies between the samples (spiked with the same cell counts) taken from different localities (Wando, Yeosu, Busan, and Uljin), which are located on the east and south coasts of Korea. Prior to DNA extraction, all environmental samples were spiked with known cell counts of culture strains.
Statistical analysis
Cell abundance was presented as mean ± standard deviations and was check for normality by the Shapiro-Wilk test and homogeneity of variance by Levene’s tests. A one-way analysis of variance (ANOVA) was performed to compare the abundance of each species of the C. marina complex between sample locations. The post-hoc pair-wise comparisons were conducted using the Scheffe test at a 5% probability level. Statistical analyses were performed using R packages stats v4.1.2 (R Core Team 2021) and lawstat v3.5 (Hui et al. 2008).
RESULTS
Phylogenetic relationship of the Chattonella marina complex
In phylogenetic trees based on the ITS and rbcL sequences, two strains of C. marina and one strain of C. marina var. ovata formed the monophyletic group (called Chattonella marina complex clade, Chamo clade) with C. marina var. antiqua supported by a substantial bootstrap value (99%) and Bayesian posterior probability of 1 (Fig. 2). C. subsalsa was positioned as the sister group of the Chamo clade with strong statistical support (1.00 / 100%). Three sequences of 61 strains belonging to the Chamo clade were almost identical, with the intraspecific divergence ranging from near-zero (ITS of C. marina) to 1.43% (CO1 of C. marina) and the inter-specific divergence ranging from 0.01% (between C. marina and C. marina var. ovata in ITS) to 1.35% (between C. marina and C. marina var. ovata in CO1) (Supplementary Table S1). These three species that belonging to the Chamo clade cannot be separated by ITS, rbcL, or CO1 sequence data. Therefore, C. marina var. ovata can be considered to be in a species complex with C. marina rather than a single species.
Validation of specific primers targeting the Chattonella marina complex
The DNA sequences, including the ITS regions of three species belonging to the C. marina complex and C. subsalsa, were aligned using Clustal Omega (Supplementary Fig. S1). The complex-specific primers were designed with an average length of 21 bases and melting temperatures of 53 and 58°C, while the estimated amplicon size was 384 base pairs (Table 1). The primer set was amplified in silico with most sequences of C. marina, C. marina var. ovata, and C. marina var. antiqua, and one of a terrestrial-originated bug, Diarsia rubi, by the NCBI Primer-BLAST tool. As a result of conventional PCR, the specificity of primers was also verified by no cross-reactivity, with 15 non-targeting phytoplankton targeted, excluding C. marina and C. marina var. ovata (Fig. 3). The presence of DNA in each PCR was confirmed by performing a PCR for a SSU rDNA, a gene commonly used to identify eukaryotic algae groups.
Determination of specific copies for estimating Chattonella abundance
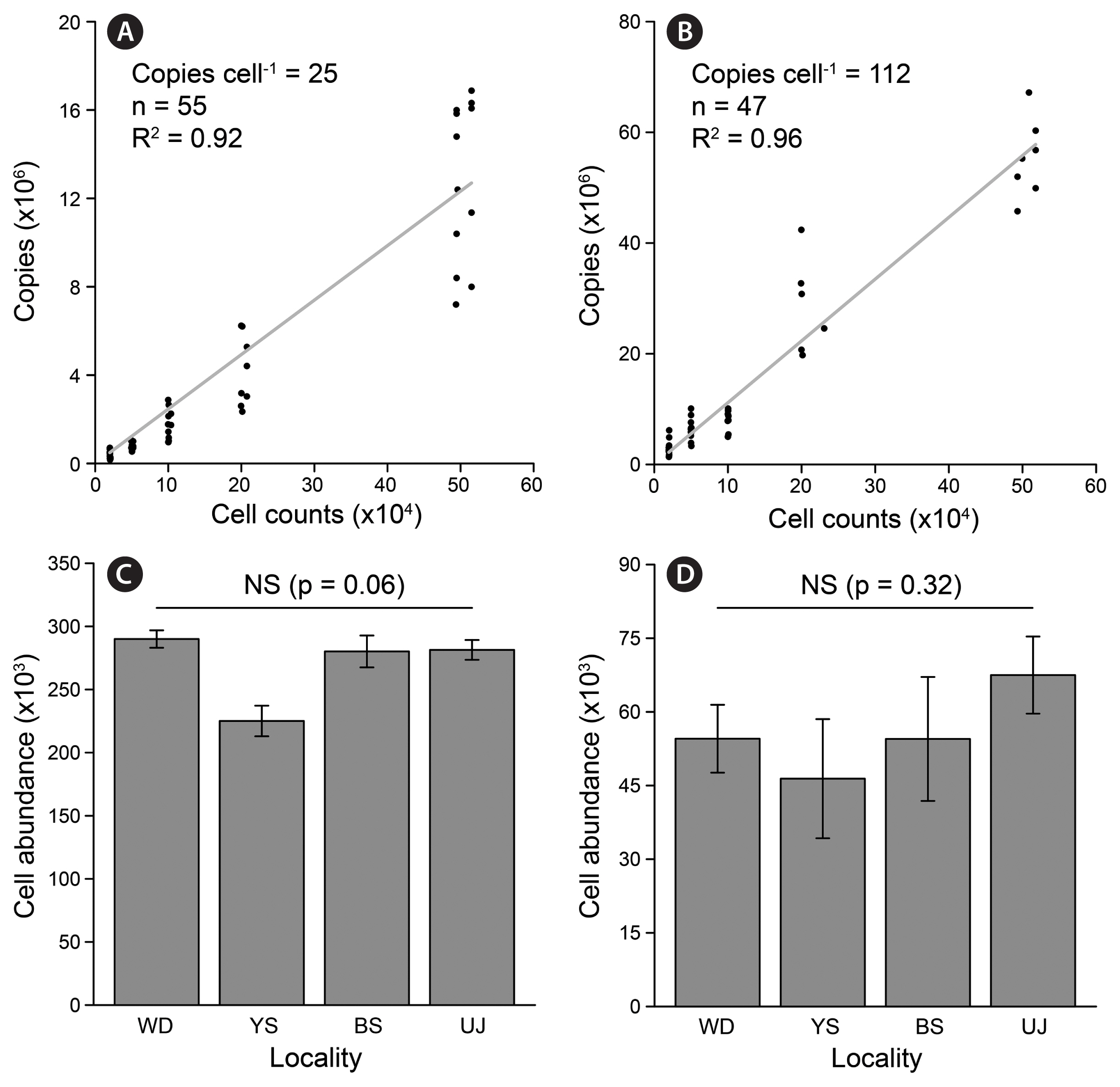
The specific copy numbers of ITS in C. marina and C. marina var. ovata were validated by the ddPCR assay based on known cell counts spiked into the Chattonella-free environmental sample (i.e., 2 × 104, 5 × 104, 1 × 105, 2 × 105, and 5 × 105 cells L−1). The total ITS copies were significantly correlated with the cell counts of C. marina (R2 = 0.92, n = 55) and C. marina var. ovata (R2 = 0.96, n = 47) (Fig. 4A & B). Although the ITS sequences of C. marina and C. marina var. ovata were identical, their copy numbers in each cell were different (25 ± 1 and 112 ± 7, respectively). To prevent saturation of accepted drops by positives, C. marina and C. marina var. ovata were spiked below 4 × 105 and 9 × 104 cells L−1 in five seawater samples collected from different locations. The results showed no significant difference at the 5% level in the cell abundance of each species between the samples taken from Korean coastal waters in the western South Sea (Wando), the South Sea (Yeosu), the eastern South Sea (Busan), and the East Sea (Uljin) (Fig. 4C & D).
DISCUSSION
The raphidophyte Chattonella species are now recognized as being frequently associated with massive fish mortality events, which have caused significant losses to commercial fish farms (Onitsuka et al. 2020). Microscopic examination of such HAB species poses a challenge when attempting to estimate their cell abundance. Tracking population dynamics of Chattonella species can be extremely difficult because their cells cannot be sufficiently preserved by commonly used fixatives such as formaldehyde, glutaraldehyde, and Lugol’s iodine (Fig. 1). There are currently no fixatives that can adequately display the representative morphological characteristics of Chattonella cells. Therefore, cell abundance can be obtained from a correlation between DNA copy numbers, which are derived from eDNA analysis with the designed specific markers, and live cell counts, which are obtained directly from the microscopic examination.
The ITS-rDNA region is widely used as a DNA barcode or genetic marker in the taxonomy and phylogeny of marine raphidophyte species because the barcode sequence is easy to amplify and it has a high nucleotide dissimilarity even between closely related species (Connell 2002, Gómez et al. 2022, Lum et al. 2022). In this study, we observed the specificity of specific primer sets, which amplified the gDNA of the target species without amplifying off-target and non-target amplicons (Fig. 3). Assays based on qPCR can be used to measure the relative abundance of target species by comparing the threshold cycles (Ct) of the target and internal control sequences. However, Ct values are strongly influenced by the efficiency of the PCR amplification, which may vary between samples depending on the amount of PCR inhibitors such as salts, exopolysaccharides, pigments, and other substances found in seawater (Schrader et al. 2012). Most qPCR assays used to enumerate cell density are based on the calibration curves, which means the abundance may depend on the accuracy of calibration and the DNA preparation yield. Meanwhile, various technical biases may affect the number of DNA copies, including DNA extraction, PCR amplification, DNA sequencing, and library construction issues (Shokralla et al. 2012). Additionally, biological biases such as biovolume and density in each cell are known to affect the gene copy number (Evans et al. 2016, Elbrecht et al. 2017). These biological biases may be universal in multicellular organisms but not in single-celled protists such as the Chattonella.
We performed a ddPCR assay without an external calibration curve or reference, in order to estimate the cell abundance of C. marina and C. marina var. ovata in marine environmental samples. It is because the cell abundance is directly calculated based on the intrinsic ITS copy number per cell of species. We systematically investigated from designing the C. marina complex-specific primers targeting the ITS region in the nucleus to applying them in order to enumerate the cells of two species. The ddPCR assay was applied for sampling, DNA preparation, running the ddPCR, and analyzing or presenting the abundance. This procedure does not need any post-normalization process to compare cell abundances, meaning that the copies themselves are enough to express how many cells are in an environmental sample (Fig. 4).
The degree of variation in ITS copies in each cell obtained from the ddPCR assay might relate to differences in the strains, intraspecific geographic location, the physiological state of the cell, or the life cycle stage (Elser et al. 2000). To prevent cell abundance being underestimation, cultured cells with similar physiological conditions and a similar cell cycle status with a decreasing number of dying cells were spiked into the samples. Indeed we confirmed that there were no significant differences in the specific ITS copies between eDNA samples from different localities, which were artificially spiked with the same cell counts (Fig. 4). The taxonomic classification of the C. marine and C. marina var. ovata remains difficult because of the degree to which they are morphologically and molecularly similar, and the existence of the species complex. Therefore, we can only attempt to quantify the C. marina complex rather than the species, considering a wide range of ITS copies in each cell from 25 to 112. A further hurdle to enumerating cells is that these species rarely dominate in phytoplankton communities in Korean waters.
In summary, the ddPCR assay provided a more rapid enumeration of C. marine complex cells, but has not allowed for the distinguishing of respective C. marine and C. marina var. ovata, where they co-exist. In this study, two methodological advantages offered by ddPCR assay should be emphasized: first, there is no need for a reference or calibration curve, and secondly, the C. marina and C. marina var. ovata in the C. marine complex are characterized by a range of ITS copy numbers. If our proposed workflow is followed, from sampling to running the ddPCR, species complex-specific ITS copies would be conserved in all environments.