INTRODUCTION
Dinoflagellates are one of the important groups comprising microbial food webs in aquatic environment and are almost constantly exposed to parasitic infections in their environment. Indeed, they are susceptible to infections by a variety of parasites and provide habitat as host for them (e.g., fungi, perkinsozoa, amoebae, dinoflagellates, euglenoids, kinetoplastids, and other heterotrophic flagellates), which spend the whole or part of their life inside or outside of their host cell, and most of the parasites cause disease and even kill the hosts (Coats 1999, Park et al. 2004, 2013, Skovgaard 2014). Among those parasites, in particular, parasitic dinoflagellates of the Amoebophrya ceratii complex, parasitic chytrid Dinomyces arenysensis, and species belonging to the family Parviluciferaceae are relatively well known, parasitoids of dinoflagellates (Norén et al. 1999, Park et al. 2004, Garcés et al. 2013, Jephcott et al. 2016, Reñé et al. 2017a). The Amoebophrya ceratii is a species complex that consists of highly host-specific to less specific parasites, with identification of species still remaining unclear so far (Coats and Park 2002, Kim 2006). The Amoebophrya infections have been reported from more than 75 dinoflagellate hosts (Park et al. 2013). This parasite has a simple life cycle consisting of three distinct stages, dinospore, trophont, and vermiform and the duration of intracellular stage, trophont, ranges from 34 to 58 h depending on the host species (Coats and Bockstahler 1994, Coats and Park 2002). The D. arenysensis is thus far an only chytrid parasite known to infect marine dinoflagellates (Lepelletier et al. 2014a). This parasitic chytrid is able to parasitize certain species of dinoflagellate in culture, with a wide host range but with a relative preference for Alexandrium species. The life cycle of the chytrid is somewhat similar to members of Parviluciferaceae, except for the development of its trophont outside host (i.e., extracellular).
Despite available numerous environmental sequences, the only Perkinsozoa genera, of which life-cycle, morphology, and ultrastructure are known, are Perkinsus, which includes species infecting molluscs, Rastrimonas subtilis, which infects freshwater cryptophytes, and Parvilucifera, parasitoids of dinoflagellates. The genus Parvilucifera has been described to contain five species, i.e., P. infectans, P. sinerae, P. prorocentri, P. rostrata, and P. corolla (Norén et al. 1999, Figueroa et al. 2008, Leander and Hoppenrath 2008, Lepelletier et al. 2014b, Reñé et al. 2017b). Very recently, two additional genera, Dinovorax and Snorkelia, parasitoids of dinoflagellates, were newly erected, with P. prorocentri being moved to the genus Snorkelia, and they were all included in the family Parviluciferaceae along with the genus Parvilucifera (Reñé et al. 2017a). The Parvilucifera species show a world-wide distribution in marine ecosystems and are known to have a broad host range (Norén et al. 2001, Park et al. 2004). By comparison, S. prorocentri is known to infect the marine benthic dinoflagellate Prorocentrum fukuyoi although its host range has not been fully investigated (Leander and Hoppenrath 2008) and host range of D. pyriformis remains unknown. Members of Parviluciferaceae have a life cycle that consists of the zoospore (free-living stage), trophocyte (feeding stage inside host cell), and sporocyte (sporogenesis stage) (Alacid et al. 2015, Jephcott et al. 2016, Reñé et al. 2017a). The free-living zoospore penetrates its host dinoflagellate and develops into a trophont while consuming the host materials. The mature sporangium contains many zoospores, and new zoospores escape from sporangium through apertures (in P. infectans, P. sinerae, P. rostrata, and P. corolla) or germ tube (= discharge tube; in D. pyriformis and Snorkelia spp.) in order to infect a new host (Norén et al. 1999, Figueroa et al. 2008, Leander and Hoppenrath 2008, Lepelletier et al. 2014b, Reñé et al. 2017a). Among the four Parvilucifera species, in particular, the two species, P. infectans and P. sinerae, are very similar or almost identical each other morphologically and genetically, thereby make it difficult to distinguish between the two. Turon et al. (2015) found that 73 clonal strains of P. sinerae, which were isolated from 10 different locations along the Atlantic and Mediterranean coats, all showed identical sequences of the small and large subunits and internal transcribed spacer of rDNA, as well as of the β-tubulin genes, although the phenotypical characterization such as zoospore success, maturation time, and infection rate showed some variability. The only main difference between the two species known so far is the number of sporangium wall (i.e., 2 layers in P. infectans vs. 3 layers in P. sinerae) (Garcés and Hoppenrath 2010). Such observations on the number of sporangium wall in those studies, however, depended on the works made on a specific time point during the sporangium development, and thus if the number of sporangium wall varies over developmental times remains unclear.
During sampling in Masan bay, Korea during the spring season of 2015, cells of the dinoflagellate Akashiwo sanguinea infected by the parasitoid Parvilucifera were observed and this host-parasite system was established in culture. The objectives of this study were to identify this Korean isolate of Parvilucifera, based on morphological (light and scanning electron microscopy), ultrastructural, and molecular data. Special attention was paid to the variation in the number of sporangium wall over developmental times. In addition, the sequences of rDNA regions (small subunit [SSU], large subunit [LSU], and internal transcribed spacer [ITS]) and β-tubulin genes were determined and compared with those of Parviluciferaceae members.
MATERIALS AND METHODS
Host and parasite cultures
The Parvilucifera infectans strain PAinf-LOHABE01 and its athecate dinoflagellate host A. sanguinea strain As-LOHABE07 were isolated from plankton samples (20 μm mesh net) taken in Masan Bay, Korea (35°12′ N, 128°34′ E) on Apr 28, 2015. The concentrated samples were examined by using an inverted microscope (Leica DM LED; Leica Microsystems, Wetzlar, Germany) to find dinoflagellates infected by P. infectans. An infected A. sanguinea cell was isolated using a drawn glass micropipette, washed six times in syringe filtered sample water (0.45 μm pore size; Advantec, Tokyo, Japan), and then transferred to a well of a 24-well plate (SPL Life Sciences, Pocheon, Korea) containing 1 mL of stock culture of A. sanguinea strain As-LOHABE03. The P. infectans was propagated by sequentially transferring either aliquots of infected A. sanguinea cells or sporangia to uninfected host culture. Stock culture of the parasite was maintained in plant culture dishes (SPL Life Sciences) and grown in 30 psu f/2-Si medium (Guillard and Ryther 1962) at 20°C under a 14 : 10 h light : dark cycle with cool-white fluorescent light providing 100 μmol photons m−2 s−1. Later, host culture of the parasite was replaced with host culture of A. sanguinea strain As-LOHABE07, which co-occurred with the parasite in the original field sample. Uninfected A. sanguinea strain As-LOHABE07 was also isolated using the same protocol described above and maintained as host stock culture under the same growth condition described above.
Light microscopy
For light microscopic observations of parasite morphology and development, A. sanguinea stock cultures in exponential growth were inoculated with recently formed zoospores and incubated for 3 d under growth conditions described above. To obtain recently formed zoospores, stock parasite culture consisting only of mature sporangia was induced to germinate by addition of syringe filtered (0.45 μm pore size; Advantec), host culture medium. Germinated zoospores accumulated near the surface of the culture dish 1 h after addition of host culture medium were harvested using a pipette. Light microscopic images of live infected host cells and mature sporangia were taken using an AxioCam HRc (Carl Zeiss Inc., Hallbergmoos, Germany) photomicrographic system coupled to an Axio Imager A2 (Carl Zeiss Inc.) equipped with differential interference contrast optics and epifluorescence capability. Images were obtained at 12, 24, 36, 49, and 70 h after inoculation. At each time, aliquots of infected host culture were fixed with 2% glutaraldehyde (final concentration), and stained with 5% SYBR Gold (final concentration) (Molecular Probes, Eugene, OR, USA) for 1 h at 4°C, and photographed using epifluorescence microscopy with blue light excitation (Filter Set 09; excitation BP 450–490, beam splitter FT 510, emission LP 515).
Scanning electron microscopy (SEM)
Cultures infected by recently formed zoospores at each time were fixed in 2% glutaraldehyde (final concentration) with 0.2 M cacodylate buffer at pH 7.4 for 1 h at 4°C. Cells were then filtered onto Isopore membrane filters (10 μm pore size for sporangia and 0.8 μm for zoospores; Millipore, Cork, Ireland), washed in distilled water for 1 h and dehydrated in a graded ethanol series (25, 50, 70, and 99%) for 12 min at each step, and then rinsed three times in absolute ethanol for each 12 min. Samples were critical point dried in liquid CO2 using a HCP-2 (Hitachi, Tokyo, Japan). Filters were subsequently glued to SEM stubs with carbon tape, sputter-coated with platinum and examined with a Hitachi FE-SEM (model S-4700; Hitachi) scanning electron microscope operating at 15 kV.
Transmission electron microscopy (TEM)
For whole mount, germinated zoospores from single sporangium into droplet were loaded onto formvar coated copper grids using a pipette. These were deposited at the gird, rinsed in distilled water, dried and examined using a JEM-1010 transmission electron microscope (JEOL, Tokyo, Japan). For TEM, aliquots of sample at each time were mixed 1 : 1 with cold 4% glutaraldehyde in f/2-Si culture medium buffered with 0.2 M cacodylate buffer at pH 7.4 at 4°C and centrifuged for 5 min at 2,000 ×g to form pellets and remove the supernatant. The samples were rinsed three times in filtered seawater, post-fixed in 1% OsO4 for 1 h at 4°C and rinsed three times in sterile deionized distilled water. Dehydration was carried out in a graded ethanol series of 50, 60, 70, 80, and 90% for 10 min each and then rinsed three times in absolute ethanol at 4°C. Pellets were then brought to room temperature and transferred through propylene oxide for 5 min, 50% Spurr’s embedding resins (Electron Microscopy Sciences, Hatfield, England) in propylene oxide for 1 h, 75% for 1 h, and 100% overnight. At the following day, pellets were transfer to new pure resin and polymerized at 70°C. Blocks were thin-sectioned on a PT-X ultramicrotome (RMC Products; Boeckeler Instruments, Tucson, AZ, USA). Sections were collected on slot grids, stained with 3% uranyl acetate and Reynold’s lead citrate, and examined using a JEM-1010 transmission electron microscope (JEOL).
The diameter of sporangium as a function of multiple infections
Recently formed (≤1 h) zoospores were added to a 24-well plate containing exponentially growing A. sanguinea host (1 × 103 mL−1) at zoospore: host ratio of 10 : 1 to induce multiple infections. After incubation of 2 d under the same growth condition described above, the diameter and number of every sporangium (n = 51) inside host were determined at ×400 magnification using an AxioCam HRc photomicrographic system coupled to an Axio Imager A2.
The number and diameter of aperture as a function of sporangium diameter
For measurement of the number and diameter of aperture in sporangium, 7-day-old empty sporangia (n = 14), which already completed the germination of zoospores, were individually picked up using a micropipette and those parameters were determined using the same photomicrographic system above at 1,000 magnification.
The number of zoospores as a function of sporangium diameter
Germination of mature sporangia was induced by the addition of syringe-filtered (0.45 μm pore size; Advantec), host culture medium. The number of zoospores that escaped from individual sporangia was determined either from video recordings of each specimen in vivo (n = 31), or direct enumeration of zoospores following fixation (n = 7). For in vivo observations, germination of single mature sporangia was recorded using an eXcope X3 digital camera (DIXI Optics, Daejeon, Korea) coupled to an inverted microscope (Leica DM LED). The zoospores were then counted while replaying the video at 0.5× normal speed. For enumeration following fixation, single mature sporangia were put into a hemocytometer and allowed to germinate. Shortly after the completion of germination, zoospores were fixed with acid Lugol’s solution and counted under an inverted microscope (BX51; Olympus, Tokyo, Japan) at ×200 or ×400 magnifications. In addition, the diameter of every sporangium was measured using eXcope X3 digital camera coupled to an inverted microscope.
DNA extraction
A single, mature parasite sporangium was isolated from stock culture using a micropipette, washed six times with sterile filtered seawater, crushed manually using the tip of the pipette, placed into a polymerase chain reaction (PCR) tube, and stored at about −80°C for 1 h. Sample was thawed at room temperature for 5 min, and DNA was extracted using Chelex 100 resin (100–200 mesh, sodium form; Bio-Rad Laboratories, Hercules, CA, USA). Fifty microliters of 10% Chelex solution was added into the PCR tube containing the sporangium and incubated at 95°C for 1 h. The tube was then centrifuged at 8,000 rpm at 4°C for 1 min. DNA present in the supernatant was transferred to a new PCR tube and stored at −20°C until analyzed.
PCR amplification and sequencing
PCR amplifications were performed with eukaryotic primers EukA and EukB (Medlin et al. 1988) for SSU rRNA gene, primers D1R and 28-1483R (Scholin et al. 1994, Daugbjerg et al. 2000) for LSU rRNA gene, primers ITS1 and ITS4 (White et al. 1990) for ITS region, and primers β-tubulinF and β-tubulinR for β-tubulin gene (Turon et al. 2015). PCRs were run in 20 μL of reaction solution containing 2 μL of DNA as a template, 2 μL of 10× Taq reaction buffer (containing 25 mM MgCl2), 0.4 μL of dNTP mix (10 mM), 2 μL of each primer (3 pmole μL−1), 0.1 μL of Taq DNA polymerase (5 U μL−1, DT16-R500; SolGent Co., Daejeon, Korea). The reactions were conducted using an automated thermocycler (C1000 Touch Thermal Cycler; Bio-Rad Laboratories) with the following conditions: for the SSU rRNA gene, an initial denaturing step at 94°C for 5 min followed by 35 cycles (95°C for 45 s, 55°C for 1 min, and 72°C for 3 min), with a final extension at 72°C for 10 min; for the LSU rRNA gene, an initial denaturing step at 95°C for 4 min followed by 40 cycles (95°C for 45 s, 52°C for 45 s, and 72°C for 2 min), with a final extension at 72°C for 7 min; for the ITS gene, an initial denaturing step at 95°C for 5 min followed by 40 cycles (95°C for 1 min, 55°C for 45 s, and 72°C for 1 min 15 s), with a final extension at 72°C for 7 min. PCR for β-tubulin were run in 20 μL of 2× TOPsimple PreMIX-HOT (Enzynomics, Daejeon, Korea) containing 5 μL of DNA as a template, 1 μL of each primer (5 pmole μL−1). PCR condition was conducted as initial denaturing step at 95°C for 10 min followed by 35 cycles (95°C for 30 s, 51.6°C for 45 s, and 72°C for 1 min), with a final extension at 72°C for 5 min. Aliquots of the amplified PCR products were electrophoresed for 25 min at 100 V in a EcoDye (SolGent Co.) stained 1% agarose gels and then visualized under UV illumination. Amplified PCR products were purified with a PCR purification kit (Bioneer, Daejeon, Korea) and sequenced with primers (EukA and EukB for SSU rRNA genes, D1R and 28-1483R for LSU rRNA gene, ITS1 and ITS4 for ITS gene, and β-tubulinF and β-tubulinR for β-tubulin gene) using a Big-Dye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems, Foster City, CA, USA) and an ABI PRISM 3730xl Analyzer (Applied Biosystems), according to manufacturer’s protocols at Macrogen Corp. (Seoul, Korea). The amplicons were sequenced until double stranded coverage was reached. ContigExpress (Vector NTI version 10.1; Invitrogen, Grand Island, NY, USA) was used to assemble the individual sequence reads and edit out low quality regions. The assembled sequences were verified by comparison using BLASTN search in the NCBI database and deposited in GenBank (accession No. MG189592 for SSU rDNA; MG189902 for ITS; MG209369 for LSU rDNA; MG229892 for β-tubulin).
Alignment and phylogenetic analyses
The obtained sequences, which are SSU, LSU, ITS, and β-tubulin region, were aligned with related sequences from GenBank database using ClustalW 1.6 (Thompson et al. 1994) and were manually refined using MacGDE 2.4 (Linton 2005). Ambiguously aligned positions were removed and final alignments of 1,499 positions for the SSU region, 815 positions for LSU region, and 2,398 positions for SSU + LSU region were selected. For β-tubulin dataset, both noncoding DNA sequences and ambiguously aligned positions were excluded, and final alignments of 660 positions were obtained. Phylogenetic trees were inferred using the maximum likelihood (ML) and Bayesian inference methods. Modeltest v.3.7 (Posada and Crandall 1998) was performed to select the most appropriate model of substitution for the ML method in PAUP. ML analyses were performed using RAxML (Stamatakis 2006) with the rapid bootstrapping option and 2,000 replicates. The GTRGAMMA evolution model was selected from MODELTEST using RAxML. TrN + I + G (−ln L = 13,743.2119), TrN + I + G (−ln L = 7115.9648), TrN + I + G (−ln L = 18996.6191), and TrNef + I + G (−ln L = 3138.8420) models were selected for SSU, LSU, SSU + LSU and β-tubulin datasets, respectively (Posada and Crandall 1998). Bayesian analysis was performed with MrBayes 3.1.1 (Ronquist et al. 2012) running four simultaneous Monte Carlo Markov Chains for 2,000,000 generations and sampling every 100 generations, following a burn in of 2,000 generations.
RESULTS
Life-cycle of the Korean isolate of Parvilucifera
The complete life-cycle and development of the Korean isolate of Parvilucifera were studies under light microscopy, SEM and TEM using A. sanguinea strain As-LOHABE07 as a host.
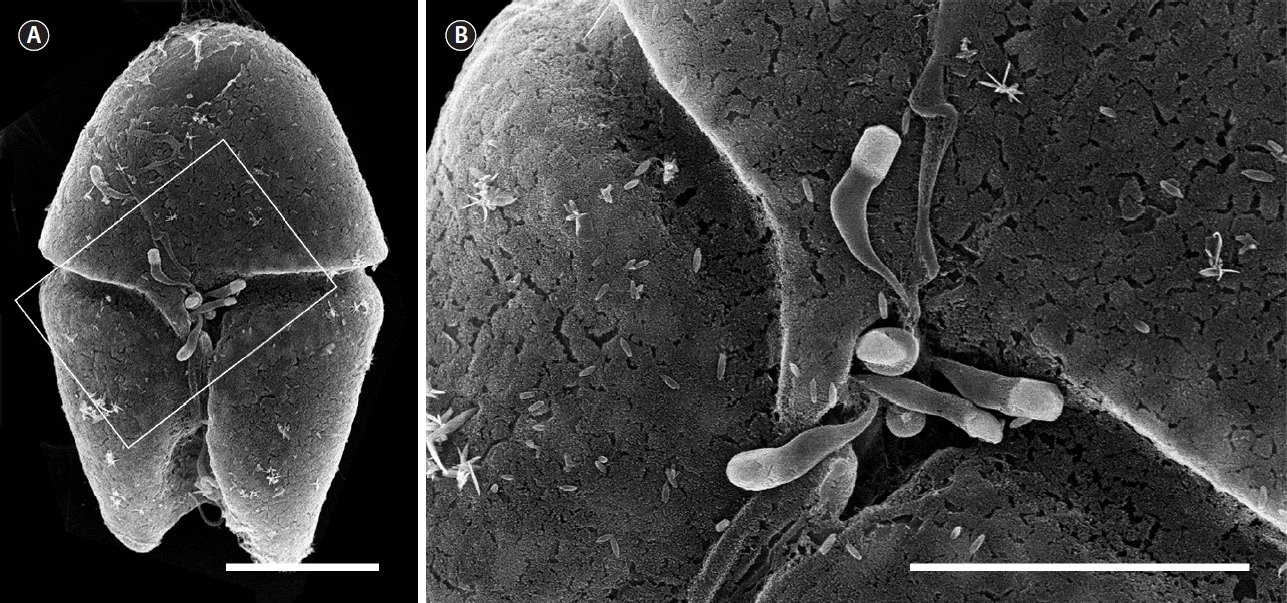
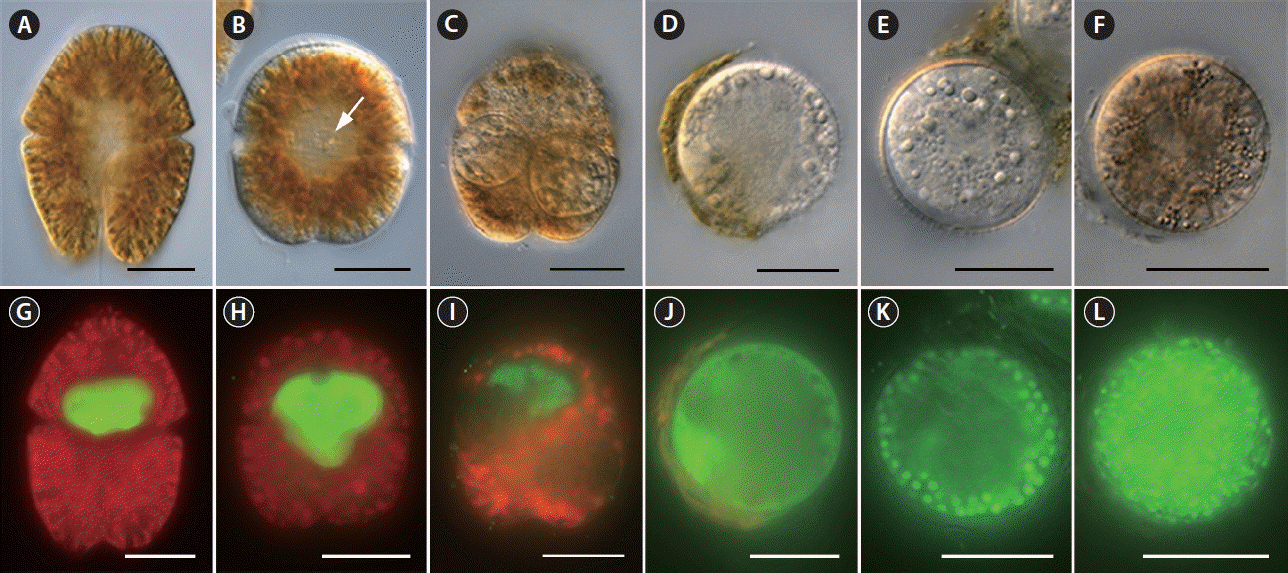
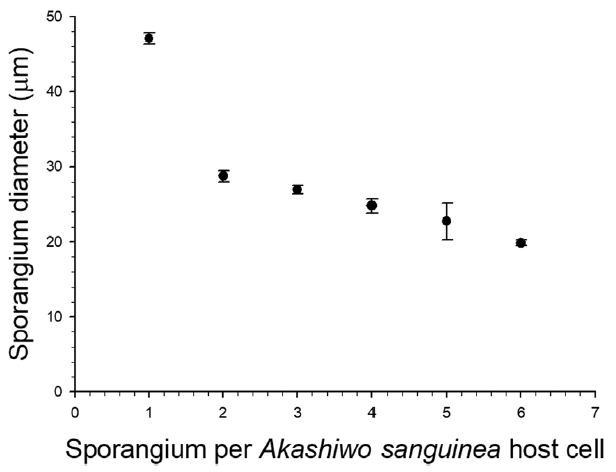
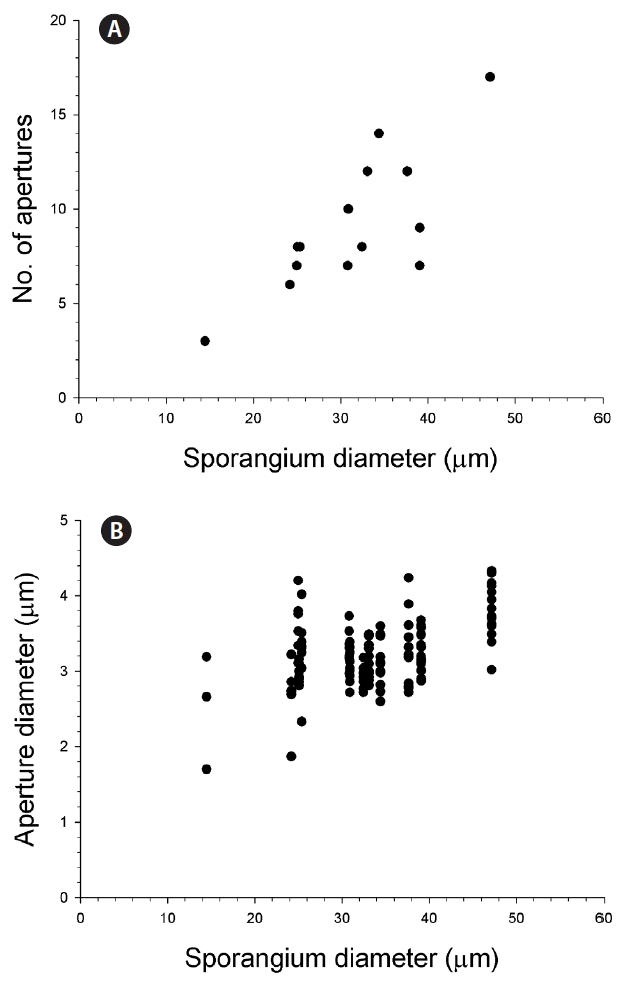
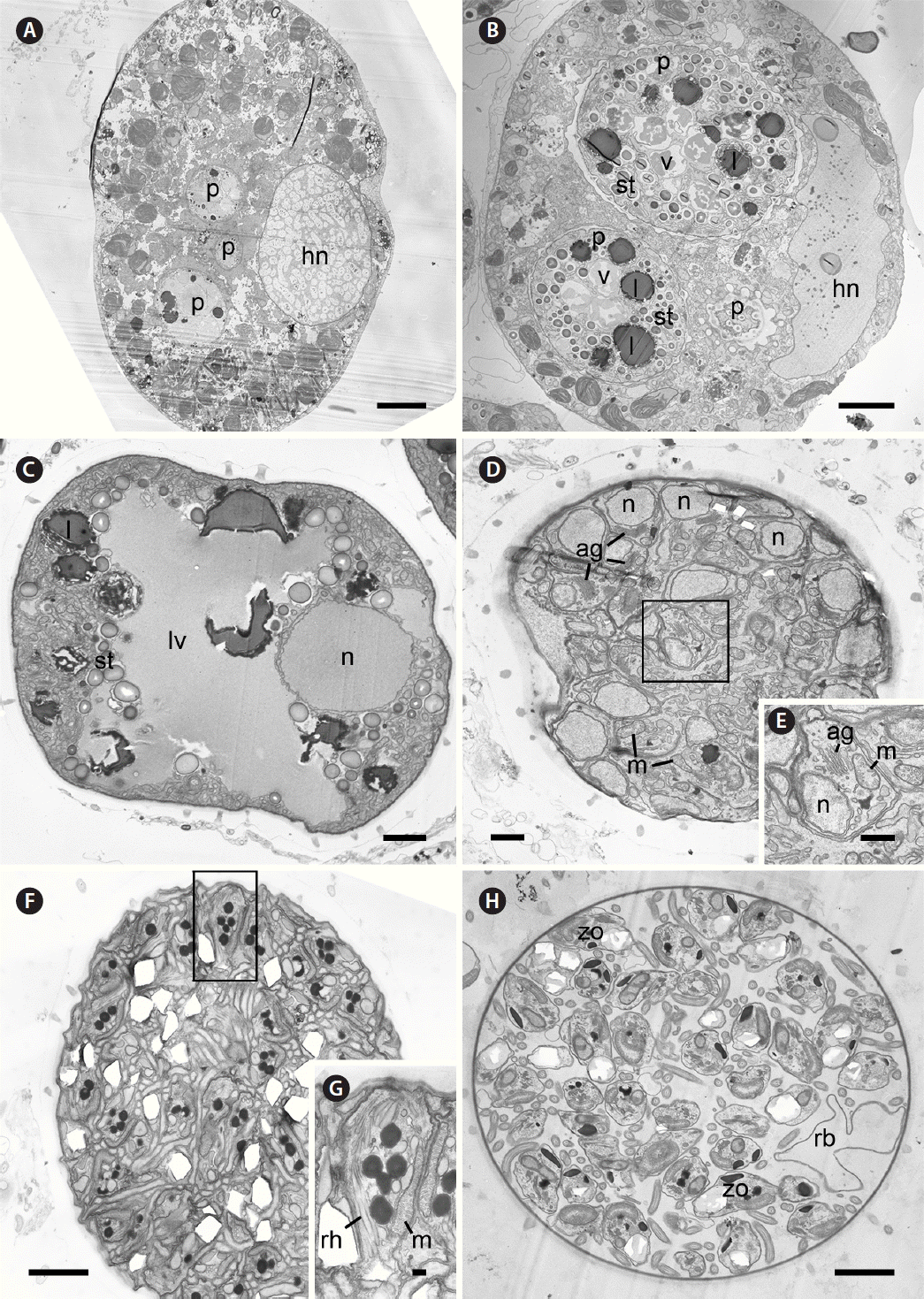
Infection occurred when a free-living zoospore penetrated into the host cell through the flagellar pore region (Fig. 1), during which the host lost its flagella and sank to the bottom of the culture flask. When exposed to zoospores, the penetration of several zoospores into a host was often observed, subsequently resulting in multiple infections (see below). During the penetration of zoospores, cell shape of the host A. sanguinea changed rapidly and became round. Early stages of infection were recognizable at 12 h after penetration by the presence of a round body in the cytoplasm of host cell (Fig. 2B). The round body continued to grow by consuming the host cytoplasm until it occupied most of the host cell and contained numerous peripheral lipid-like structures (Fig. 2C). At early trophocyte, it was hard to clear observe the nucleus of parasite when stained SYBR gold (Fig. 2I), due probably to the shortage of condensed genetic materials. At the late trophocyte, sporangium was released from the host, which was almost completely consumed (Fig. 2D), and at this time nucleus of the parasite was invisible when stained with SYBR gold (Fig. 2J). At the early sporocyte (49 h), a large vacuole was observed in the center of the sporangium and peripheral site was filled with zoospores as clearly seen in SYBR gold-stained sporangium (Fig. 2E & K). During the late stage of infection (70 h), the sporangium was full of numerous zoospores and the mature sporangium was dark-colored under the light microscope (Fig. 2F & L). Under the growth conditions described above, it took 3 days from addition of Parvilucifera zoospores to A. sanguinea to development of mature sporangia. In a single A. sanguinea cell, even up to 9 sporangia were observed to develop. When more than 1 sporangium developed, their size decreased with increasing number of sporangia (Fig. 3). The sporangium diameter ranged from 12.3 μm in case of multiple infections to 50.4 μm in single infection. When the zoospores were fully developed and ready to be escaped, they swam within the sporangium and escaped from the sporangium through several apertures. The number and diameter of aperture appeared to be dependent on sporangium diameter (Fig. 4). The number of aperture ranged from 3 to 17. The aperture was round-shaped and measured from 1.7 to 4.3 μm in diameter. Each aperture was covered by an operculum until release of the zoospores. The number of zoospores produced per sporangium was related with sporangium size (Fig. 5). When infecting A. sanguinea, minimum and maximum numbers of zoospores produced were 34 and 2,300 zoospores per sporangium, respectively.
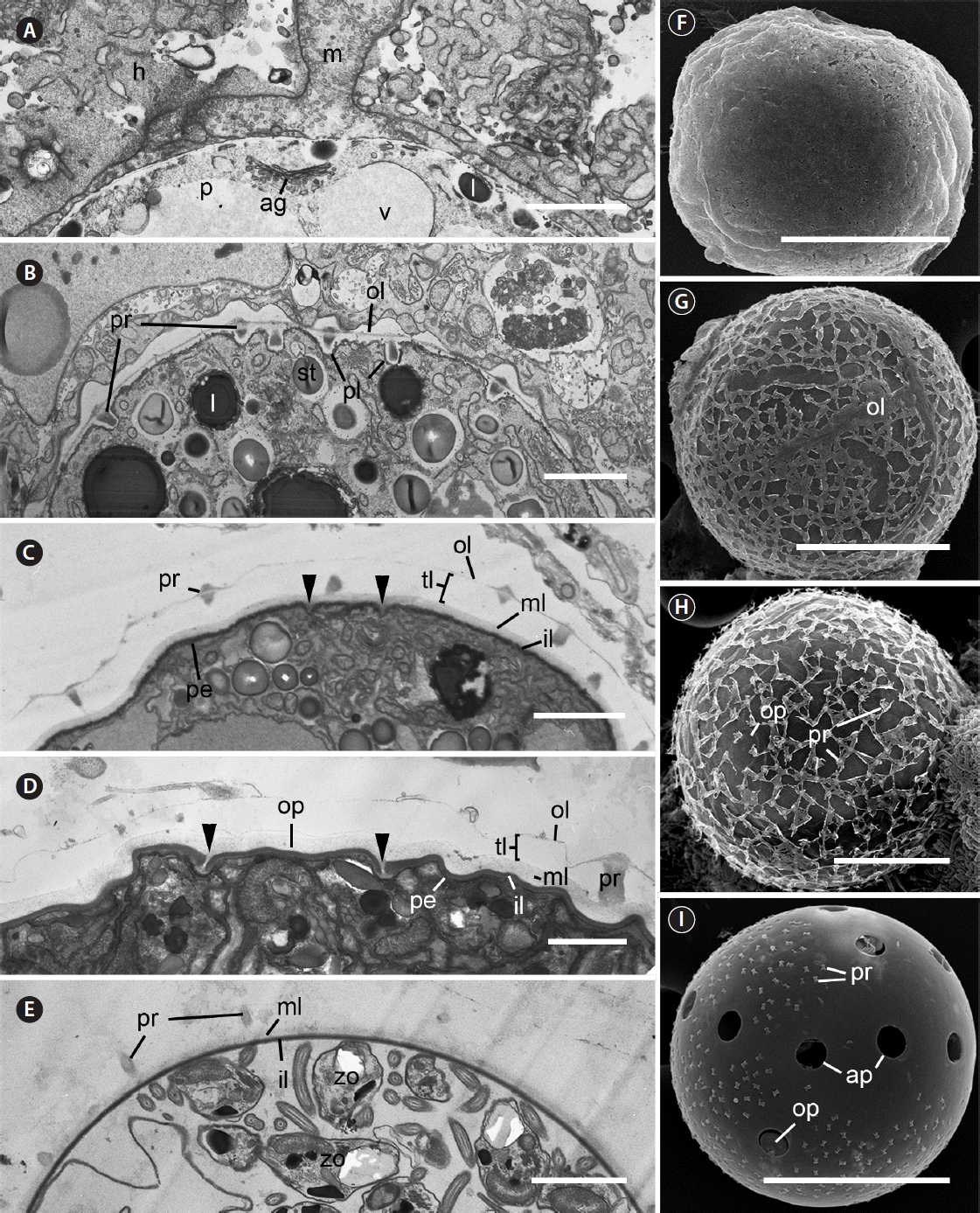
Ultrastructure of trophocytes and sporocytes and development of the sporangium wall
Early trophocyte was visible inside the host cytoplasm 12 h after inoculation of zoospores (Fig. 6A). Early trophocyte grew inside the parasitophorous vacuole and contained vacuoles, a Golgi apparatus, and lipid inclusions. The mitochondria of the host were frequently observed outside parasitophorous vacuole (Fig. 7A). The trophocyte gradually grew, increasing in size while consuming the host cytoplasm and contained vacuoles, starch granules, and lipid globules at 24 h (Fig. 6B). At this time, the parasitoid plasma membrane with folded surface was visible and was covered with parasitophorous vacuole membrane. Developing processes were also found, embedding between parasitoid plasma membrane and parasitophorous vacuole membrane (Fig. 7B). At 36 h, the trophocyte occupied most part of the host body and sometimes released from the completely disintegrated host. The trophocyte was characteristic of the presence of a large vacuole in the central part, together with several starch granules, lipid globules, and a large nucleus (Fig. 6C). The outer layer was still connected to the distal ends of processes. At this time, sporangium wall was observed to be clearly consisting of four-layers (i.e., innermost, middle, transparent, and outer layers). The transparent layer became more evident at 36 h, compared to that at 24 h. Invagination of the pellicle underneath innermost layer started to occur, probably for formation of operculum (Fig. 7C). At this stage, the released sporangium was characterized by covering with an intact outer layer as well as remnant of host materials (Fig. 7F). At 49 h, the division of nuclei and cellular organelles was observed (Fig. 6D & E). The outer layer of sporangium started to degrade at this time (Fig. 7G). At 70 h, sporangium was filled with numerous immature zoospores and pellicles were observed between zoospores (Fig. 6F & G). The innermost layer was cut off to form operculum (Fig. 7D). At this time, the outer layer continued to degrade and it was easy to observe processes on the surface of sporangium (Fig. 7H). Mature sporangium was filled with fully developed zoospores and residual body (Fig. 6H). At 1-week-old sporangium, the outer layer fully disappeared and even processes were detached (Fig. 7E & I).
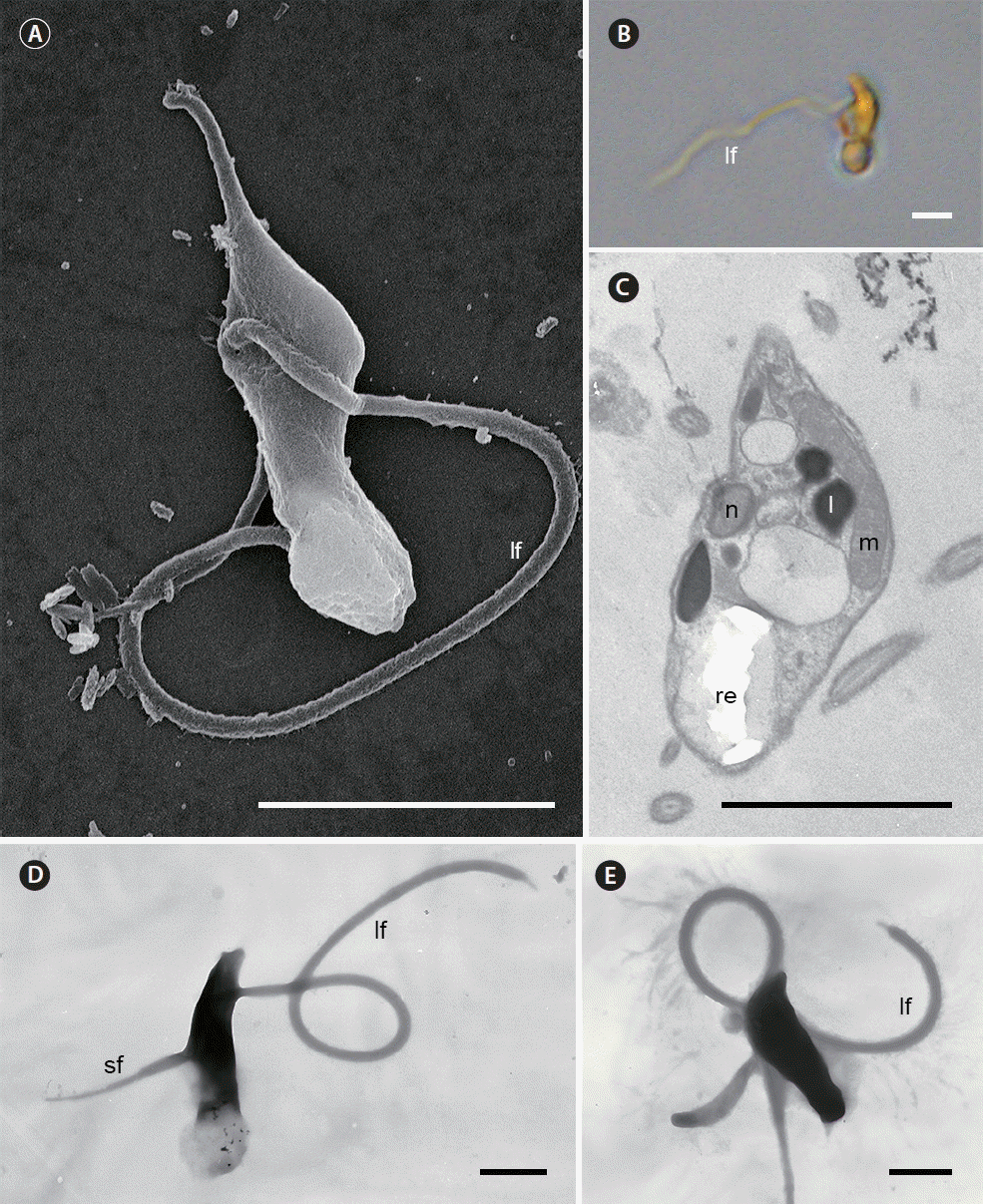
Zoospore
The zooids just germinated from sporangium had an elongated body (5.31 ± 0.35 μm in length, 1.38 ± 0.09 μm in width, n = 14 using SEM). Swimming zoospores, which had a large refractile body and a long flagellum, were easily visible under the light microscope (Fig. 8B). SEM and whole mounts clearly revealed the presence of two flagella of unequal size (Fig. 8A, D & E). The short flagellum measured 2.79 ± 0.19 μm (n = 7), was bare, and had a proximal swelling (Fig. 8D). The short flagellum was located quite close to the cell body and was difficult to observe under light microscope. The long flagellum measured 12.77 ± 0.67 μm (n = 3) and terminated in a short conical tip (Fig. 8D & E). The long flagellum had hairs (Fig. 8E), but its hairs were rarely visible in the whole mounts, unlike the absence in SEM. The two flagella were located at near the central part of the body (Fig. 8A & D). Zoospore had a large mitochondrion and an elongated nucleus at ventral side in the middle of the cell. A large refractile body was located at the posterior end of zoospore and lipid globules were observed in zoospore (Fig. 8C).
Molecular phylogeny
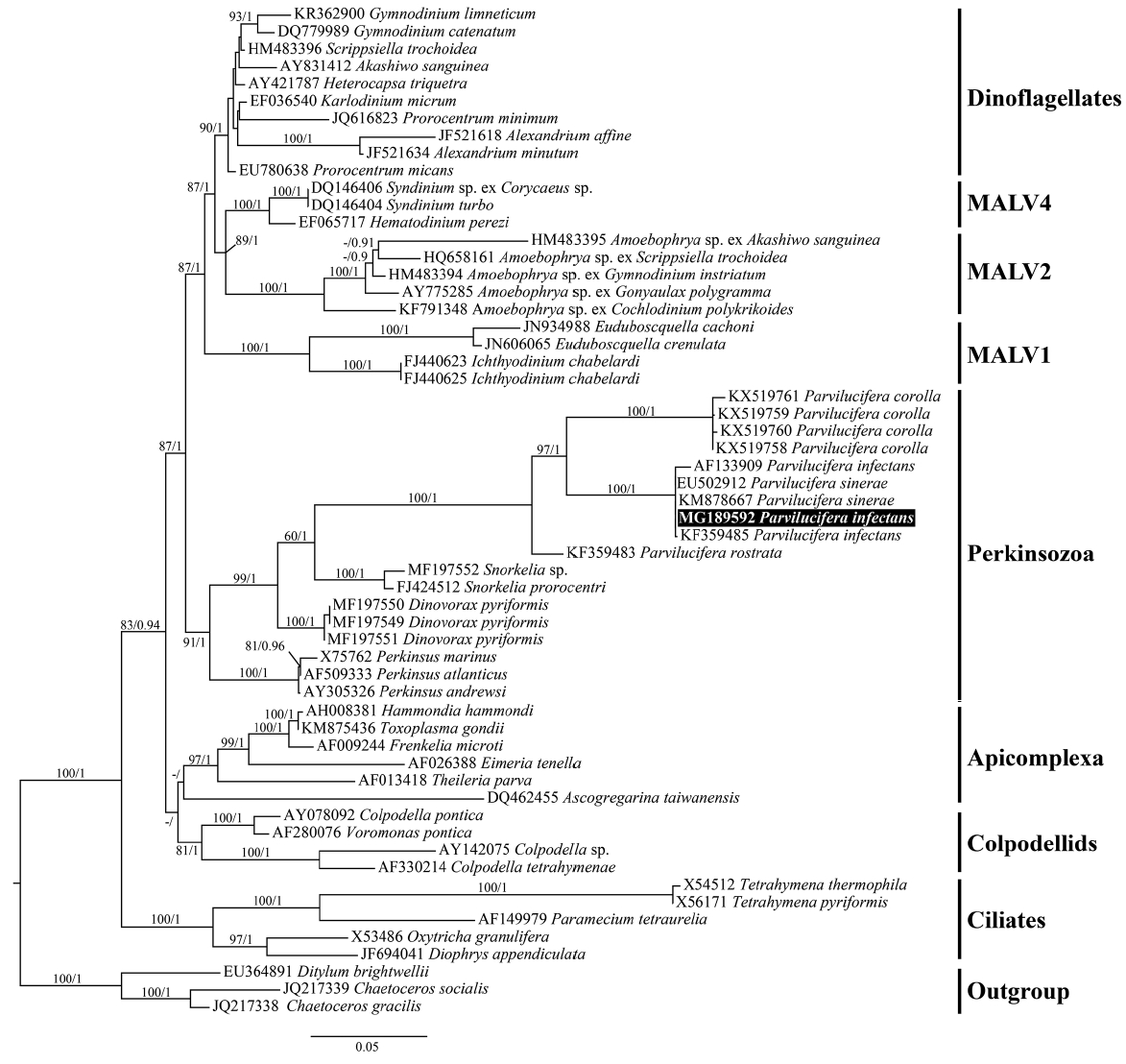
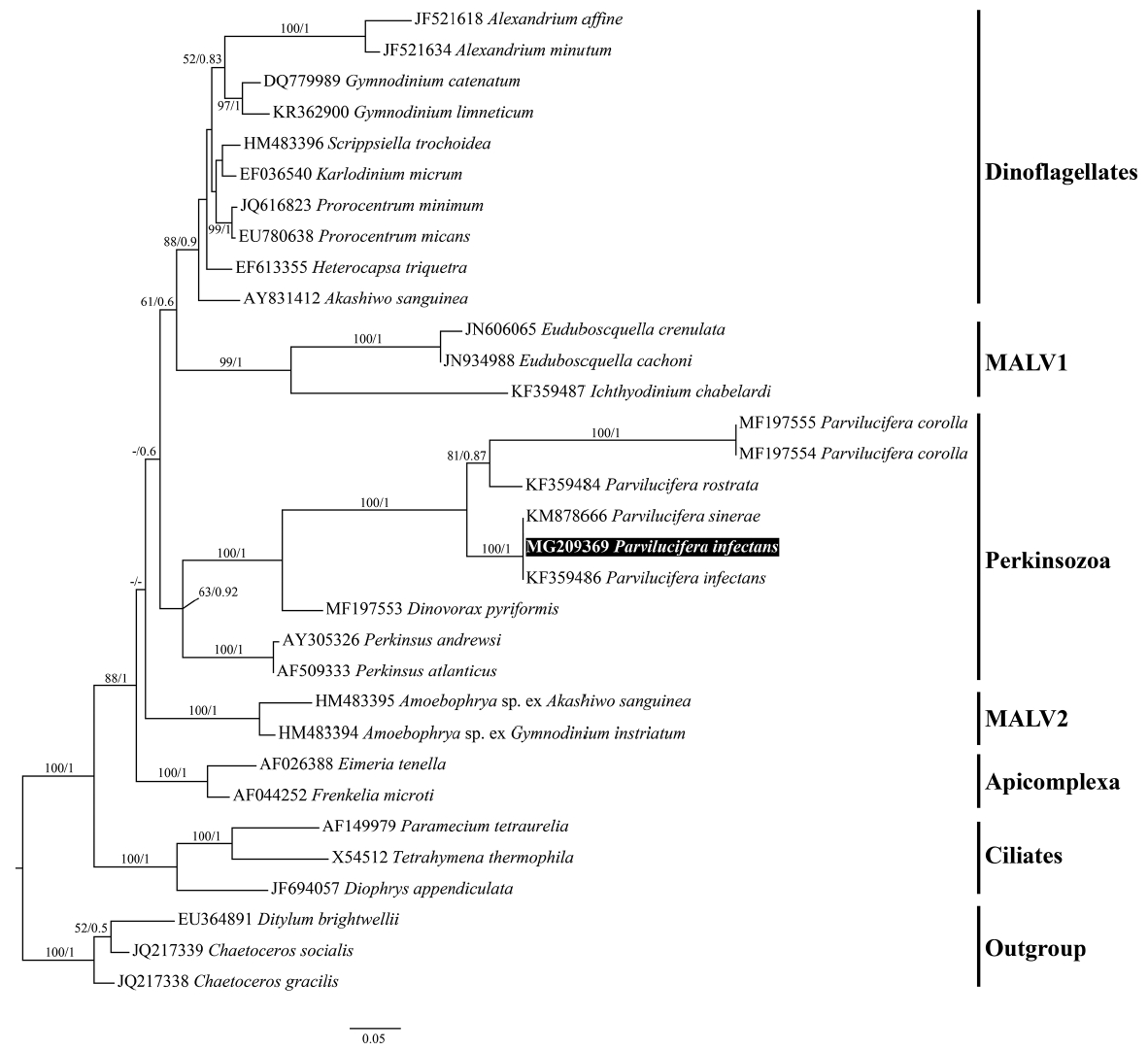
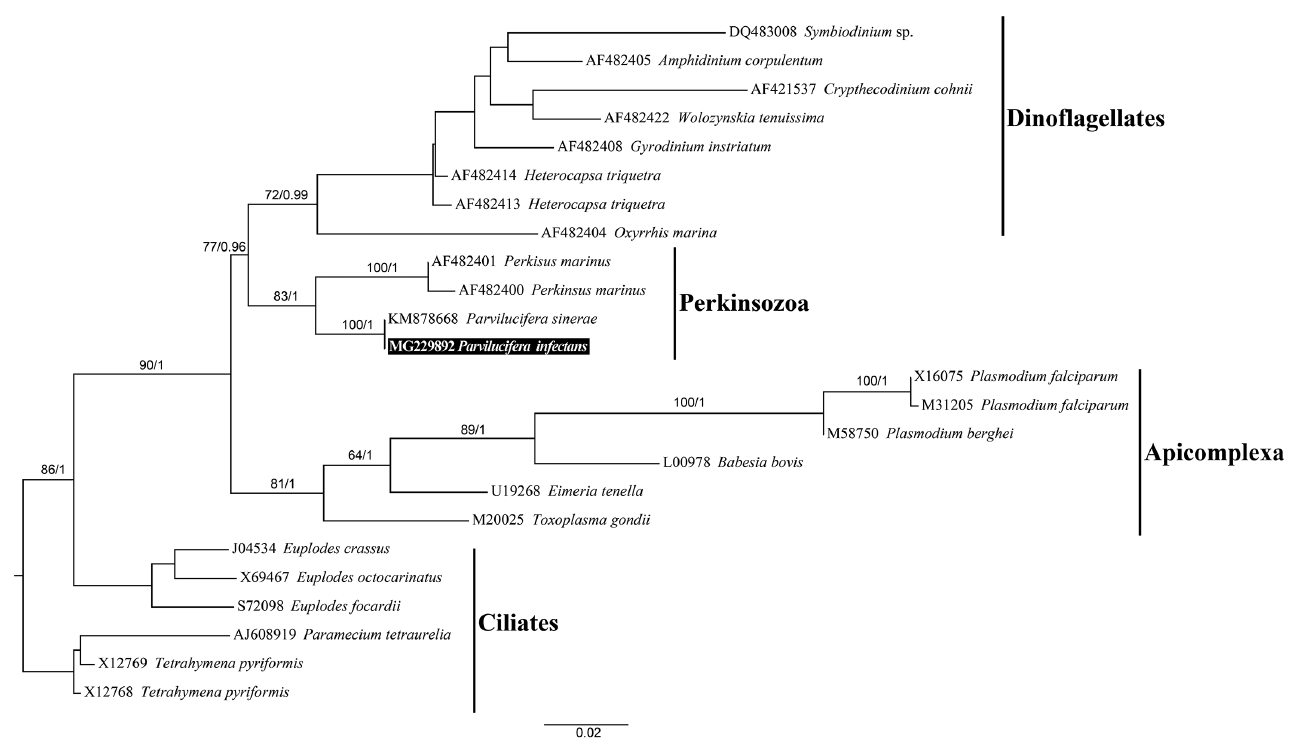
From 10 individually isolated single sporangium, partial SSU rDNA sequences of 1,421 bp, partial ITS rDNA sequences of 463 bp, partial LSU rDNA sequences of 1,176 bp, and β-tubulin sequences of 957 bp (319 amino acids) were obtained, with 100% identity among the parasites. Phylogenies based on the SSU rDNA (Fig. 9), LSU rDNA (Fig. 10), SSU rDNA + LSU rDNA (Supplementary Fig. S1), and β-tubulin (Fig. 11) gene sequences provided overall congruent tree topologies that coincided with the ones already published for those genes (Figueroa et al. 2008, Lepelletier et al. 2014b, Turon et al. 2015, Reñé et al. 2017a, 2017b) although phylogenetic position among Parvilucifera species changed in the LSU rDNA phylogeny. The Korean isolate of Parvilucifera was included in a clade that contained the sequences of P. infectans / P. sinerae species complex in all phylogenies, with a high bootstrap support (ML, 100%) or a posterior probabilities (PP) of 1. In all trees, all members of Parviluciferaceae (Parvilucifera, Snorkelia, and Dinovorax) known so far and Perkinsus species clustered together as a clade corresponding to Perkinsozoa, but had low statistical supports. In the case of β-tubulin, the Korean isolate grouped with the sequence of P. sinerae, with strong bootstrap support (ML, 100%) or a PP of 1 (Fig. 11).
The percentage of genetic similarities between the rDNA and β-tubulin sequences of Korean isolate of Parvilucifera and those of members of Parviluciferaceae is shown in Table 1. The SSU rDNA sequence (AF133909) of P. infectans in GenBank deposited by Norén et al. (1999) had 100% identity with that of P. infectans specimen collected at the same location (Kristineberg Marine Research Station, Sweden) as the type species (F. Norén personal communication). The SSU rDNA sequence of the Korean isolate had 99.1% similarity (23 nucleotides difference) with those of P. infectans (AF133909) reported by Norén et al. (1999) as well as P. infectans specimen from type locality and 99.5% similarity (7 nucleotides difference) with the P. infectans sequence (KF359485) reported by Lepelletier et al. (2014b), but it was 100% identical to the P. sinerae sequence (KM878667) reported by Turon et al. (2015). The SSU rDNA sequence of the Korean isolate, however, differed from those of P. rostrata (KF359483), P. corolla (KX519760), Snorkelia prorocentri (FJ424512), and Dinovorax pyriformis (MF197551), with 87.7, 85.7, 80.3, and 80.5% similarities, respectively. The LSU rDNA sequence of the Korean isolate was all 100% identical to those of P. infectans (KF359486), P. infectans specimen collected from the same area as type locality (F. Norén personal communication), and P. sinerae (KM878666), but it differed from the sequences of P. rostrata (KF359484) (68% similarity), P. corolla (MF197555) (49.9% similarity), and D. pyriformis (MF197553) (49% similarity). The ITS region of the Korean isolate sequence had 100% similarity with the sequences of both P. infectans (KF359485) published by Lepelletier et al. (2014b) and P. sinerae (KM878665) published by Turon et al. (2015), but had 54% similarity with the sequence of P. rostrata (KF359483). In the case of β-tubulin, the sequence of the Korean isolate was 100% identical to that of P. sinerae (KM878668) reported by Turon et al. (2015).
DISCUSSION
Following the first description of P. infectans by Norén et al. (1999) almost two decades ago, species diversity in the family Parviluciferaceae greatly increased recently, reaching up to 6 species across three genera (Figueroa et al. 2008, Leander and Hoppenrath 2008, Lepelletier et al. 2014b, Reñé et al. 2017a, 2017b). Among the members of Parviluciferaceae, species belonging to genera Dinovorax and Snorkelia can be easily distinguishable from those of the genus Parvilucifera even simply by some morphological features of sporangium. For example, sporangium of D. pyriformis and S. prorocentri can be distinctly distinguishable from other Parvilucifera species by the presence of a germ tube, which can also be seen in another genus Perkinsus within the Perkinsid (Azevedo 1989), and its characteristic smooth surface without processes (Leander and Hoppenrath 2008, Reñé et al. 2017a). By comparison, the sporangia from Parvilucifera species (P. infectans, P. sinerae, P. rostrata, and P. corolla) have some similarities in morphology in that they commonly possess numerous processes and variable number of operculae at the surface (Norén et al. 1999, Figueroa et al. 2008, Lepelletier et al. 2014b, Reñé et al. 2017b). Lepelletier et al. (2014b) reported that the processes of P. rostrata are shorter than those of P. infectans, suggesting that the difference in length may be a key feature for identifying the Parvilucifera species. The range (0.44–0.76 μm) in length of processes from a Korean isolate of Parvilucifera, however, overlapped with that (0.55–0.65 and 0.4–0.45 μm, respectively) of P. infectans and P. rostrata, suggesting that the difference in processes lengths is not a good character for identifying the Parvilucifera species. Despite several morphological similarities, however, species delineation among P. infectans / P. sinerae, P. rostrata, and P. corolla seems to be easily made based on molecular data such as differences in SSU rDNA sequence (see below). At present, the most problematic case is found in the two species, P. infectans and P. sinerae, in which they are very similar or almost identical morphologically and genetically.
The Korean isolate of Parvilucifera investigated in the present study more closely resembled P. infectans / P. sinerae species complex rather than P. rostrata and P. corolla morphologically and ultrastructurally. Garcés and Hoppenrath (2010) reported through an ultrastructural study that the sporangium wall consists of different number of layers between P. infectans and P. sinerae: while the former has sporangium wall consisting of two layers, the latter has sporangium wall consisting of three layers. Based on this difference, they argued that the difference in numbers in layer of sporangium wall can distinguish between the morphologically very similar two Parvilucifera species. As shown in this study, however, the number of layers in sporangium wall can be variable even in the same species, depending on which layers are included or not and the age of sporangium examined to count the layers (Table 2). Previous ultrastructural studies (e.g., Norén et al. 1999, Lepelletier et al. 2014b, Alacid et al. 2015) have not considered the presence of a transparent layer, although it was clearly visible between middle layer and outer layer in their original figures, but if this transparent layer may be considered as a layer, then the number in layers of sporangium wall would increase to even four. In the present study, the transparent layer between middle layer (i.e., corresponding to opaque layer in Norén et al. 1999; less opaque but thicker middle layer in Garcés and Hoppenrath 2010; thick and loose layer in Lepelletier et al. 2014b; medium layer in Alacid et al. 2015) and outermost layer (i.e., connected between processes; corresponding to thin outer non-membrane layer in Lepelletier et al. 2014b; outer layer in Alacid et al. 2015) was included in counting the number of layers of sporangium wall, thereby making the number of layer of sporangium wall from Korean Parvilucifera isolate characterized by a total of 4 layers. Given this criterion about layer of sporangium wall, sporangium wall from P. infectans and P. sinerae all seems to be consisted of 4 layers (Table 2). For example, closer examination of Fig. 9 in the original paper by Norén et al. (1999) revealed that the sporangium wall in P. infectans appears to consist of 4 layers, in which the third transparent and fourth outermost layers could be clearly visible but were not considered by the authors. While Garcés and Hoppenrath (2010) counted and included innermost, middle, and transparent layers for sporangium wall of P. sinerae, Alacid et al. (2015) considered innermost, middle, and outer layers for the same species, without including the transparent layer.
In addition to layers to be considered for counts, the number in layers of sporangium wall can change over time. The present study clearly demonstrated that the trophocyte at 36 h was covered with four layers, and then outer layer of the sporocyte gradually degraded over time, resulting in wall structure consisting of three layers, with even processes being detached from 7-day-old sporangium with smooth surface. Similar to this, Lepelletier et al. (2014b) also reported gradual degradation of the outer layer with time. The disappearance of the outer layer with increasing times seems to be associated with the release of fully developed zoospores along with the opening of operculum in mature sporangium. Taken together, the difference in the number of layers seems not to be an appropriate ultrastructural character for sorting Parvilucifera species (especially P. infectans and P. sinerae), unlike the suggestion by Garcés and Hoppenrath (2010).
In Parvilucifera species, zoospores of free-living stage have generally rather elongated body in shape and two flagella consisting of hairy long flagellum and bare short flagellum, with the exception of S. prorocentri, in which information about hairs has not reported yet (Table 2). Closer examination of the published data, however, raises the question of whether the body shape of zoospore (in particular, from P. sinerae) is really elongated. For example, Figueroa et al. (2008) reported that zoospores of P. sinerae have elongated body in their text, but zoospore from SEM images presented in their paper measured about 2.7 μm in length and 2.2 μm in width, indicating quite a rounded shape of zoospore. Also, closer examination of Fig. 6 in Garcés and Hoppenrath (2010) and Fig. 4G in Alacid et al. (2015) revealed that elongated zoospores exist in sporangium from TEM images, and body shape of these zoospores is very similar to P. infectans and P. rostrata, which have elongated body reported by Norén et al. (1999) and Lepelletier et al. (2014b). Taken together, a possibility that the SEM images presented by Figueroa et al. (2008) might be resulted from the contaminated organism in culture cannot be excluded. On the other hand, Norén et al. (1999) mentioned that hairs of long flagellum were sometimes visible in whole mounts. In the present study, the hairs of zoospore from Korean isolate were also rarely visible in whole mounts, and even were never visible in SEM images. The reason for this remains uncertain at present, but the possibilities that either hairs may be detached from the flagellum during the fixing process or Parvilucifera may produce two types of zoospores cannot be excluded. The formation of different types of spores has already been reported in syndinean parasites of the ciliates such as Euduboscquella spp. (e.g., Coats et al. 2012). Further study needs to examine zoospores of all Parvilucifera species to test if they produce two types of zoospores.
Phylogenetic analysis also supported that the Korean isolate of Parvilucifera is closer to P. infectans / P. sinerae species complex rather than P. rostrata and P. corolla. The LSU rDNA sequence of the Korean isolate was 100% identical to that of P. infectans specimen collected at the same location as the type species (F. Norén personal communication), as well as to those of P. infectans (Norén et al. 1999, Lepelletier et al. 2014b) and P. sinerae (Turon et al. 2015). Unlike LSU rDNA sequence, however, some differences in the SSU rDNA sequences within P. infectans / P. sinerae species complex were found. When not included 2 ambiguous bases in the SSU rDNA sequences of P. infectans (Norén et al. 1999, Norén personal communication), they differed in 19–21 nucleotides (1.1–1.4% differences) from those of the Korean isolate of Parvilucifera, P. infectans (Lepelletier et al. 2014b), and P. sinerae (Turon et al. 2015). Nonetheless, it should be noted that the SSU rDNA sequences of P. infectans by Norén and his colleagues contain 9 unusual nucleotides, in which the Alveolata shares highly conserved, common nucleotides, and thus which seem to be sequencing errors. Thus, when excluded both 2 ambiguous and 9 unusual nucleotides, SSU rDNA sequences of P. infectans (Norén et al. 1999, Norén personal communication) differed in at most only 11–13 nucleotides (0.6–0.9% differences) from those of other researchers, including the Korean isolate. Given the differences in 190–272 nucleotides (12–16% differences) between P. infectans / P. sinerae species complex and other Parvilucifera species (i.e., P. rostrata and P. corolla), therefore, such small differences seem not to be large enough to discriminate between the two species, P. infectans and P. sinerae. Further, those small differences in SSU rDNA sequences of P. infectans / P. sinerae species complex may reflect the variations among different strains of the same species from different geographical areas. A good example for this case can be found in P. corolla. For example, even if two isolates of P. corolla were collected from the same Canary Island (North Atlantic), their SSU rDNA sequences (KX519760 and KX519761) differed in 10 nucleotides (0.6% difference), comparable to the level found in P. infectans / P. sinerae species complex mentioned above.
In conclusions, the morphological and ultrastructural observations of the Korean isolate of Parvilucifera established in culture highlighted the inconsistency of using the number of layers from sporangium wall, which has been believed as a morphological character to discriminate between P. infectans and P. sinerae (Garcés and Hoppenrath 2010). The present study revealed that the number of layers from sporangium wall depends on the stage of sporangium development and the layers considered for counts. While pairwise comparison of the LSU rDNA sequences showed 100% identity among P. infectans / P. sinerae species complex, genetic differences were found in the SSU rDNA sequences but the differences were very small (11–13 nucleotides) compared with those (190–272 nucleotides) found among the rest of Parvilucifera species (P. rostrata and P. corolla). Those small differences in SSU rDNA sequences of P. infectans / P. sinerae species complex may reflect the variations within inter-strains from different geographical areas. Taken together, all morphological and ultrastructural characteristics and molecular data suggest that P. infectans and P. sinerae are the same species. Based on all morphological and ultrastructural characteristics and molecular data, the Korean isolate of Parvilucifera fits P. infectans (Norén et al. 1999), which is the older name with priority.