An advanced tool, droplet digital PCR (ddPCR), for absolute quantification of the red-tide dinoflagellate, Cochlodinium polykrikoides Margalef (Dinophyceae)
Article information
Abstract
To quantify the abundance of the harmful marine dinoflagellate Cochlodinium polykrikoides in natural seawaters, we developed an innovative procedure based on the droplet digital PCR (ddPCR) with C. polykrikoides-specific primers targeting the internal transcribed spacer (ITS). The abundance of C. polykrikoides was determined through the specific copy number of target DNA per cell in both culture and field samples. The average copy number per C. polykrikoides cell as determined via the ddPCR was 157 with using known cell numbers and a simplified protocol for preparing DNAs. C. polykrikoides cell numbers, in samples collected from different locations, calculated via ddPCR were in agreement with direct cell counts using a microscope. The ddPCR protocol was then used to measure the abundance of C. polykrikoides in nearshore and offshore waters of the southern coast of Korea. Practical application of this method demonstrated its usefulness in reducing time spent on analysis and for increasing estimate accuracy.
INTRODUCTION
Red tides or harmful algal blooms (HABs) can cause large-scale fish mortality and consequently may have substantial economic effects on the aquaculture industry (Park et al. 2013). In response, countries around the world have invested considerable sums toward exploring new approaches for reducing losses resulting from HAB. Monitoring of the species responsible for HABs is a critical step in managing and minimizing losses in the aquaculture sector (Sellner et al. 2003, Park et al. 2013, McKibben et al. 2015).A variety of dinoflagellates are known to cause HABs in the world’s oceans (Richlen et al. 2010, Kudela and Gobler 2012, Lee et al. 2013, Jeong et al. 2016); of these, the dinoflagellate Cochlodinium polykrikoides is one of the primary sources of HAB events, and thus a leading cause of mass mortality of farmed fish and shellfish in many countries (Shahraki et al. 2013, 2014, Rountos et al. 2014). To reduce the economic and environmental impacts of C. polykrikoides, identifying and measuring its cell abundance prior to a bloom are critical, but identifying C. polykrikoides based solely on its morphological features is often difficult because of its similarity to other Cochlodinium species that occur in Korean waters (Cho et al. 2001, Matsuoka et al. 2008, Iwataki et al. 2010, Marcoval et al. 2013). In addition, learning to identify HAB species under a microscope often requires long period of training for inexperienced researchers, and is a time-consuming, labor-intensive, and error-prone process even for experts. As such, it is vital that methods of quickly and accurately identifying and quantifying the abundance of HAB species be developed.
Pigments can be useful biomarkers for detecting particular phytoplankton groups, but are often inadequate for identifying a particular species when phylogenetically related species are also present (Jeffrey and Vesk 1997). Bioactive metabolites and toxins are also frequently used as biomarkers in environmental monitoring programs along with plastid or mitochondrial DNA, such as rbcL, tufA, and COI genes, but their quantities are often dependent on the physiological status of the species (Bereiter-Hahn 1990, Sato et al. 2003, Park et al. 2016). Genomic DNA (gDNA) found in the nucleus of eukaryotic organisms represents the complete set of genetic information of an organism, and exhibits features that differ in size and complexity among species (Coleman and Mai 1997, Buchheim et al. 2011, Hong et al. 2016). Although gDNA sequences are not available for all marine species, the sequences of several ribosomal DNA (rDNA) regions of numerous marine species have been sequenced for use in phylogenetic analyses. In addition, these regions are used for designing species-specific primers that can be used to identify and enumerate species via polymerase chain reaction (PCR)-based tools. Two conditions must be met to ensure accurate quantification of HABs using PCR with species-specific primers in the environmental samples: samples used for both the gDNA extraction process and the PCR tool used in quantifying the amount of target gDNA must be free of naturally-occurring contaminants, such as dirt or salts.
Quantitative real-time PCR (qPCR) analysis is the most common method used for identifying and monitoring HAB species. However, it can also serve as a tool for measuring the relative amount of species with fold changes in a sample based on the internal and external controls, or by simply comparing threshold cycles (Ct) to predefined standard Ct levels that are generated using a known number of cells (Coyne et al. 2005, Park et al. 2014, Lee et al. 2017). Although standard curves offer reliable estimates of the abundance of particular species in complex marine communities, qPCR analysis may not generate accurate representations of cell densities because Ct is highly sensitive to PCR inhibitors in seawater, such as salts, pigments, exopolysaccharides, humic acids, and other substances (Flekna et al. 2007, Ellison et al. 2011), reinforcing the need for the development of new molecular approaches.
A major advantage of droplet digital PCR (ddPCR) over qPCR is that it is relatively insensitive to the existence of PCR inhibitors in environmental samples, and thus optimizes reproducibility and consistency when attempting to estimate amount of target DNA fragments. Moreover, standard curves or references used to determine the number of target DNAs or for interpreting the abundance of specific species are not required in ddPCR (Sanders et al. 2011, Scollo et al. 2016). For accurate estimation of DNA copy number using ddPCR, the target DNA fragments must be randomly distributed across a large number of partitions, and 20,000 nanoliter droplets in each ddPCR reaction and the absolute copy number of target DNA should be calculated statistically based on the ratio of droplets carrying target DNA fragments. The ddPCR process therefore provides estimates of the abundance of a species with greater speed, accuracy, and sensitivity than do microscopic observation or qPCR assays.
In this study, we constructed a C. polykrikoides-specific primer by using the sequences of the internal transcribed spacer (ITS) region between 18S and 28S rDNA. Using the primer and the ddPCR analysis, we quantified the abundances of C. polykrikoides in coastal waters of the southern Korea in 2016 and 2017. We believe that the results of this study provide a solid foundation for the use of a ddPCR approach in the early detection and monitoring of harmful C. polykrikoides blooms.
MATERIALS AND METHODS
Isolation, culturing, and sampling
Cultures of Cochlodinium polykrikoides collected from Yeosu Bay in southern coast of Korea were grown at 23°C in enriched f/2 seawater media under ~300 µmol photons m-2 s-1 illumination and a 12 h : 12 h light : dark cycle using four 36 W daylight fluorescent lamps (Dulux L 36W/865; Osram, Münich, Germany). The cultures were transferred to new media on a weekly basis.
To validate inter-sample variability and the consistency of the ddPCR assay, we collected various seawater samples from different locations along the southern coast of Korea (Muan, 35.02° N, 126.37° E; Wando, 34.31° N, 126.76° E; Taean, 36.62° N, 126.35° E; Gunsan, 35.93° N, 126.52° E) in spring and summer 2016. In addition, we used ddPCR to determine the abundance of C. polykrikoides in samples collected from nearshore and offshore waters of the southern coast of Korea during cruises of the National Institute of Fisheries Science (NIFS) research vessel Tamgu 20 in August 2016 and 2017. Seawater samples (l L) were filtered through a 0.45 µm cellulose acetate (CA) filter (Advantech Inc. Tokyo, Japan) and stored at -80°C until gDNA was prepared for use in quantifying the specific copies per sample volume by ddPCR. In 2016, C. polykrikoides occurrence can only be determined by inverted microcopy on Lugol-preserved samples that were prepared by netting, whereas in 2017, Lugol-preserved field samples (1 L) were concentrated by 1/10 using the settling and siphoning method and quantified through a microscope using a Sedgwick-Rafter counting chamber.
Overall procedure
A flow chart illustrating the overall procedure for quantifying C. polykrikoides using ddPCR is shown in Fig. 1. Briefly, the culture sample containing known cell numbers was filtered through a 0.45 µm CA filter to collect C. polykrikoides cells, and field seawater samples in which its abundances were unknown were also filtered in the same way under low vacuum (<100 mm Hg). C. polykrikoides gDNA was prepared from the filter using simple steps, as explained next in the DNA preparation section; gDNA in the homogenized supernatant was analyzed as the template for each PCR. Approximately 1 h of homogenization was needed to process 24 samples simultaneously, which is a more consistent and faster process than the affinitycolumn-based methods that are common to commercial DNA prep kits for plants or soil (Qiagen, Hilden, Germany). A glass-fiber filter (GFF; Millipore, Billerica, MA, USA) was also used to collect cells to compare the effectiveness of different filter media, but we found that GFFs yielded very low amounts of gDNA, and moreover the copy number for each C. polykrikoides fluctuated greatly because of the affinity of GFF to negatively-charged nucleic acids.

Schematic diagrams of genomic DNA (gDNA) preparation and droplet digital polymerase chain reaction (ddPCR) analysis for estimating the abundance of target species. (A) Seawater samples containing known and unknown densities of Cochlodinium polykrikoides were filtered through a cellulose acetate filter at low vacuum. (B) gDNA (as a PCR template) was prepared by lysis with beading and boiling. (C) ddPCR was performed using 5 µL of the gDNA prepared in (B) and determination of the specific copy number of target DNA was based on the C. polykrikoides with the internal transcribed spacer (ITS)-specific primer set.
DNA preparation
Initial concentrations of C. polykrikoides were established using an autopipette to deliver a predetermined volume of culture with a known cell number to the bottles containing seawater collected from the different locations, and gDNA used for determining the copy number per C. polykrikoides cell was extracted on a CA filtering. Field seawater samples were also filtered for the same purpose under relatively low vacuum. Extraction of gDNA from both the culture samples with known cell numbers and the field samples required a two step process: first, cells were disrupted on a filter with five 2 mm Zirconia beads (Watson Co., Tokyo, Japan) in a lysis buffer (25 mM NaOH and 2 mM EDTA), and second, via boiling in a ThermoMixer C (Eppendorf AG, Hamburg, Germany) for 30 min. Following neutralization through the addition of an equal volume of 40 mM Tris-HCl (pH 5.5), 5 µL of the gDNA supernatant solution was used as the template for each PCR.
PCR and droplet digital PCR
A C. polykrikoides specific primer (C.poly ITS-F, 5′-GACGCAGCGAAGTGTGATAA-3′ and C.poly ITS-R, 5′-CAACGCCTTGACAAACAAGA-3′) was designed based on the ITS sequences, the specificity of which was validated using the NCBI Primer-BLAST tool (https://www.ncbi.nlm.nih.gov/tools/primer-blast/), and also against regular PCR purified gDNA from C. polykrikoides, C. fulvescens, Alexandrium spp., Dinophysis spp. (dinoflagellates), Pseudo-nitzschia sp. (diatom), and Ulva ohnoi (green alga); no cross-reactivity was observed (supplementary Fig. S1). Ribosomal DNA (rDNA), which is widely used in molecular phylogenic studies, has highly conserved sequences that are organized as a repeated multigene family separated by an ITS in the genome. Sequences of Dinophyceae ITS (located between 18S and 28S rDNA) were obtained from the NCBI nucleotide bank (https://www.ncbi.nlm.nih.gov/nuccore/) and aligned with the ITS sequences of C. polykrikoides to identify variable sequence regions that could be used to construct C. polykrikoides specific primers. Thermal cycling consisted of 30 cycles of denaturation at 95°C for 30 s, annealing at 58°C for 20 s, and extension at 72°C for 20 s in a T100 Thermal Cycler (Bio-Rad Lab Inc., Hercules, CA, USA); 20 µL of PCR reaction was used in total, along with 10 µL of 2× prime Taq premix (Genet Bio, Daejeon, Korea) and 1 µL of 2 pmol of each primer (forward and reverse) with 0.1 µg DNA templates.
A QX200 Droplet Digital PCR (ddPCR) system (Bio-Rad Lab Inc.) was used to quantify the absolute copy numbers per C. polykrikoides cell. We followed the manufacturer’s recommended protocols for droplet generation and for reading the droplets after running the same PCR conditions. Each ddPCR reaction was performed in a final volume of 20 µL containing 10 µL of EvaGreen Supermix (Bio-Rad Lab Inc., Munich, Germany), 1 µL of forward and reverse primer (2 pmol each), and 5 µL of the gDNA solution. Following PCR amplification, the droplets were read with a QX200 Droplet Reader and QuantaSoft software ver. 1.7.4 (Bio-Rad Lab Inc., USA), which counts the number of positive- or negative-drops used to calculate the absolute quantification of the target DNA. All chemicals used in the ddPCR assay were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA) or Bio-Rad (Bio-Rad Lab Inc., USA), unless otherwise specified.
Data analysis
Data are presented as mean ± standard error. One-way analysis of variance (ANOVA) followed by Duncan’s multiple comparison tests, or a Student-Newman-Keuls test for multiple comparisons in Minitab 14 (Minitab Inc., State College, PA, USA), were used to compare the number of copies per C. polykrikoides cell between the cell densities, sample locations, and ddPCR measures and microscopic cell counts.
RESULTS AND DISCUSSION
Here, we presented an advanced quantifying method based on ddPCR analysis with C. polykrikoides-specific primers. To determine the copy number of target DNA per C. polykrikoides cell, ddPCR was performed on a gDNA soup prepared from culture samples containing known cell numbers (Fig. 2). Data with over 10,000 drops in total, along with a threshold obtained by adding 5,000 to a basal amplitude for eliminating false positives, were included in estimations of the specific copy number per C. polykrikoides cell (Fig. 2A & B). Total copies of C. polykrikoides were highly linearly correlated with cell number (r2 = 0.930, n = 56), and no significant difference in the average number of copies per cell was detected between cell number (p = 0.609). A single cell of C. polykrikoides produced an average of 157 ± 16 copies of the target DNA fragments (Fig. 2C).
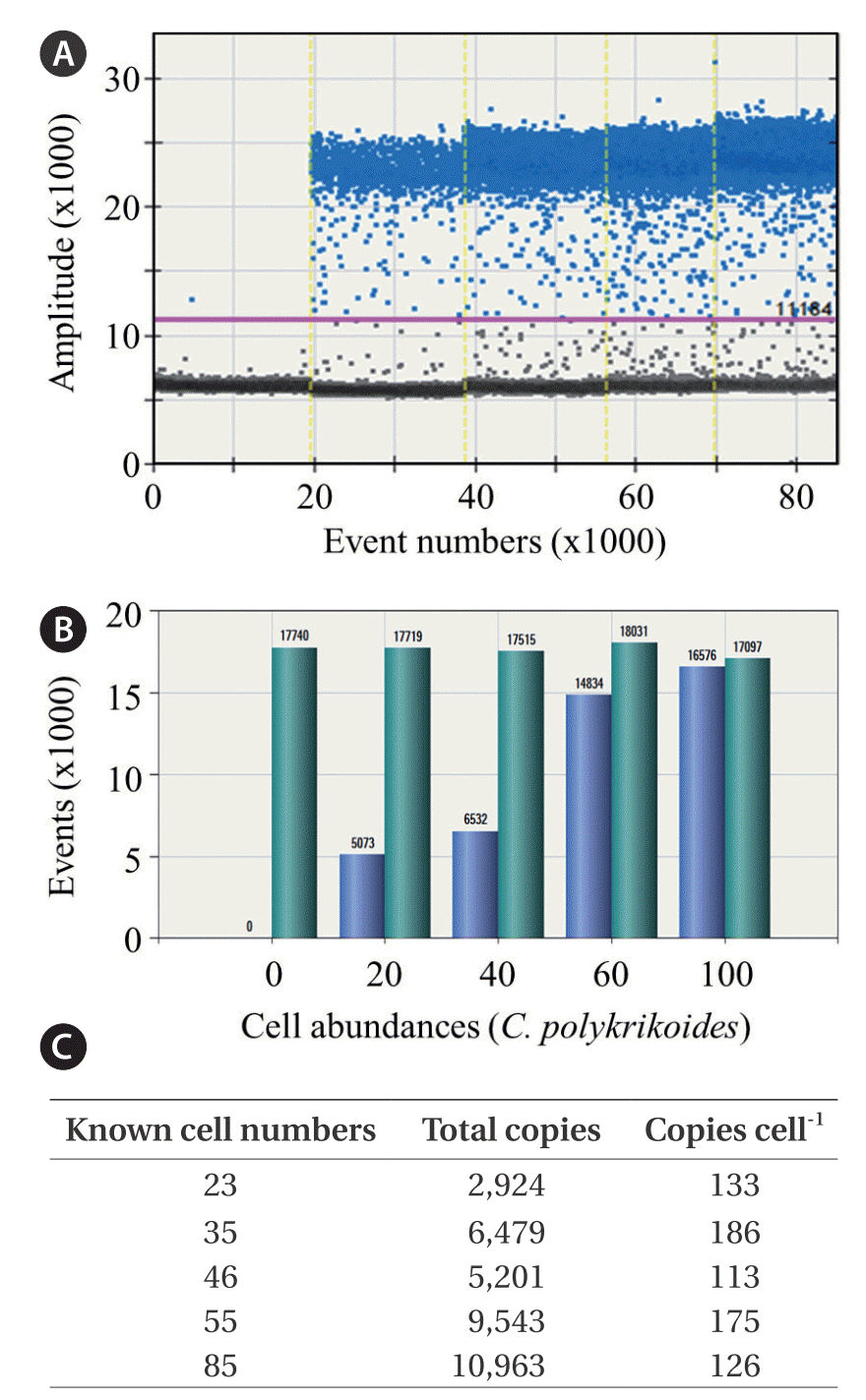
Droplet digital polymerase chain reaction (ddPCR) was performed with genomic DNA (gDNA) of Cochlodinium polykrikoides to determine the specific copy numbers of target DNA sequences with the internal transcribed spacer (ITS)-specific primer. (A) Total and positive signaled droplets were visualized after running ddPCR, and data with over 10,000 droplets in total were included in the calculation of the copy number. (B) Calculation of the specific copy number was based on the threshold by adding 5,000 at the basal negative amplitude. (C) To acquire the genomic copy number per C. polykrikoides cell, the ddPCR was performed on culture samples containing various known cell numbers.
The cell abundances determined using ddPCR were compared to those by direct microscopic cell counts (Fig. 3A). Although ddPCR values were slightly higher than those derived from direct microscopic cell counts, the difference between the two methods was not significant (p = 0.221, n = 3). Moreover, the specific copy number per single cell measured by ddPCR did not differ among the various seawater samples collected from different locations (p = 0.395, n = 3) (Fig. 3B). Taken together, these results suggest that in addition to facilitating detection of C. polykrikoides even when it occurs at very low abundances, ddPCR is a cost- and time-effective tool for monitoring HAB species, and thus represents a significantly more efficient approach for evaluating field samples from marine environments that contain complex mixtures of organic and inorganic material.

Comparison of Cochlodinium polykrikoides abundance as determined by droplet digital polymerase chain reaction (ddPCR) and microscopic cell counts (A), compared between seawater samples collected at different locations (B), which contained prepared genomic DNA (gDNA) (approximately 100 cells). NS, not significant at p > 0.05.
Following verification of the ddPCR method as appropriate for quantifying C. polykrikoides in seawater, we applied this method to samples collected in nearshore and offshore waters of the southern coast of Korea. In August 2016, C. polykrikoides was detected in samples taken from eight of 25 stations, with abundances exceeding 100 cells L−1 at only one station (400-17) (Fig. 4). To test the detection of C. polykrikoides by ddPCR at the eight stations, Lugol’s fixed samples prepared by netting at the same time as sampling was undertaken were observed under a microscope, and C. polykrikoides cells were observed at only three stations. In August 2017, seawater samples for quantitative evaluation by both microscopic cell counts and ddPCR assays were collected at five separate locations (Table 1). The abundance of C. polykrikoides was counted only in samples taken from Masan, for which an average of 320 cells L−1 were detected by microscopic observation and 193 cells L−1 by ddPCR assay. No C. polykrikoides cells were observed under the microscope in samples collected from the other four locations, whereas ddPCR detected C. polykrikoides at two locations, Mokpo and Goheung. These results suggest that ddPCR can be used to quantify the abundance of target species even when they occur in very low densities.
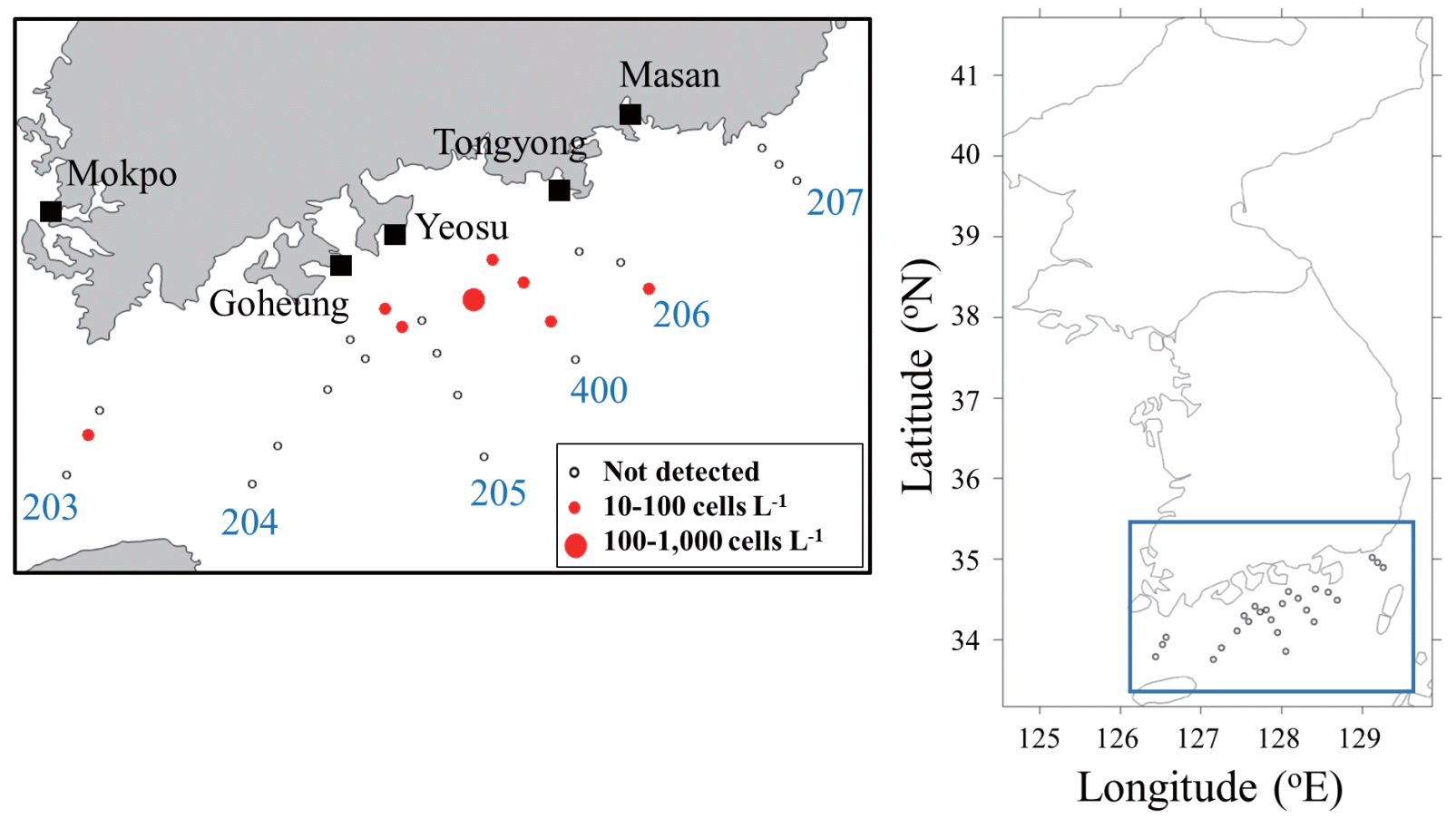
Map of sampling stations and cell abundance of Cochlodinium polykrikoides determined by droplet digital polymerase chain reaction (ddPCR) off the southern coast of Korea in late August 2016.

Comparison of microscopic observation and ddPCR assay of Cochlodinium polykrikoides cell abundance in Korean coastal waters in August 2017
There are several possible reasons for the discrepancy between the two methods of quantifying abundances, including the relatively low densities of C. polykrikoides that occurred in the South Sea of Korea during the summers of 2016 and 2017, which may have been insufficient for visual detection of C. polykrikoides cells. Since the 1980s, C. polykrikoides blooms have reappeared annually, but no blooming events were reported along the Korean coastline in either 2016 or 2017. In addition, an unarmored chain-forming Cochlodinium that exhibits a wide range of morphological variability was found to easily rupture and pass through the net mesh, thereby confounding C. polykrikoides identification. Moreover, identifying cysts through microscopic observation is often unreliable because cysts can be extremely difficult to recognize (Kim et al. 2007), and thus discerning cysts through a microscope may be an inadequate approach for identifying species in the early stages of bloom formation.
PCR technologies, regular PCR, qPCR, and dPCR, are highly sensitive tools for analyzing the occurrence and amount of target DNA with species-specific primers. qPCR visualizes a relative amount of target DNA by comparing its threshold cycles (Ct values) directly, or by comparing Ct values with those of control or reference groups. Regular PCR and qPCR are less effective because of the presence of PCR inhibitors in seawater samples, however, which may result in inaccurate estimates of target DNA; one major advantage of ddPCR is that, unlike qPCR, it can tolerate the presence of PCR inhibitors in samples, which helps to maintain reproducibility and consistency. Moreover, identifying gDNA copies via standard curves or references is not required for ddPCR assays (Sanders et al. 2011, Devonshire et al. 2015, Scollo et al. 2016).
Cochlodinium polykrikoides blooms have become more frequent in recent decades as a results of global warming and increasing eutrophication, and consequently their impacts on fisheries and aquaculture industries have intensified worldwide (Kudela and Gobler 2012, Lee et al. 2013, Park et al. 2013). Regular monitoring, including the rapid and precise identification and quantification of red-tide causing dinoflagellate species, is needed to predict when and where these blooms occur. We believe that application of ddPCR represents a more efficient means of quantifying the absolute abundance of C. polykrikoides in the marine environment.
CONCLUSION
The copy number of ITS fragments per cell is unique to each species and thus can be used to determine the abundance of C. polykrikoides even in complex environments. In this study, we developed a novel procedure for preparing gDNA and performing ddPCR to quantify the abundance of red-tide causing C. polykrikoides. Some modifications can be made to improve the accuracy and sensitivity of monitoring C. polykrikoides abundance with our ddPCR protocol; for instance, specific RNA-target primers can be used to exclude possible gDNA contamination from non-living C. polykrikoides cells, and more than one specific primer set or multiple markers for a particular species can be used to improve the accuracy of estimates, although this would require more detailed information about the genome and transcriptome of the targeted species. How methods for the early detection and monitoring of invasive and harmful organisms in marine ecosystems—including but not limited to C. polykrikoides—can be improved will be the focus of future research.
SUPPLEMENTARY MATERIAL
Validation of Cochlodinium polykrikoides-specific primer. (A) NCBI Primer-BLAST tool demonstrated no cross-reactivity with other species against C. polykrikoides-specific primers. (B) Electrophoresis after performing regular polymerase chain reaction (PCR) with C. polykrikoides-specific primers indicated no PCR amplification product against genomic DNA of other non-target species (https://www.e-algae.org).
ACKNOWLEDGEMENTS
Financial support for this research was provided by the “Management of Marine Organisms Causing Ecological Disturbance and Harmful Effects” program (KIMST/MOF) and NRF-2016R1A6A1A03012647 to KYK; and the Basic Science Research Program of the NRF, funded by the Ministry of Science and ICT (NRF-2015R1C1A1A02037499) to HGL.
Abbreviations
CA
cellulose acetate
ddPCR
droplet digital polymerase chain reaction
gDNA
genomic DNA
GFF
glass-fiber filter
HAB
harmful algal bloom
ITS
internal transcribed spacer
PCR
polymerase chain reaction
qPCR
quantitative real-time polymerase chain reaction